
The Problem With Beta-Carbolines, Part III
Some armchair-testable predictions about psychotic disorders

Earlier this year, I wrote about the etiology of schizophrenia, positing that psychotic disorders are caused by an overabundance of gut bacteria which produce the psychoactive chemicals tryptamine and phenethylamine.
The genesis of the hypothesis lay in an Australian study from 2024, which used shotgun metagenomics to look at the gut microbiome in schizophrenia, and found some absolutely huge differences between patients and mentally healthy controls:
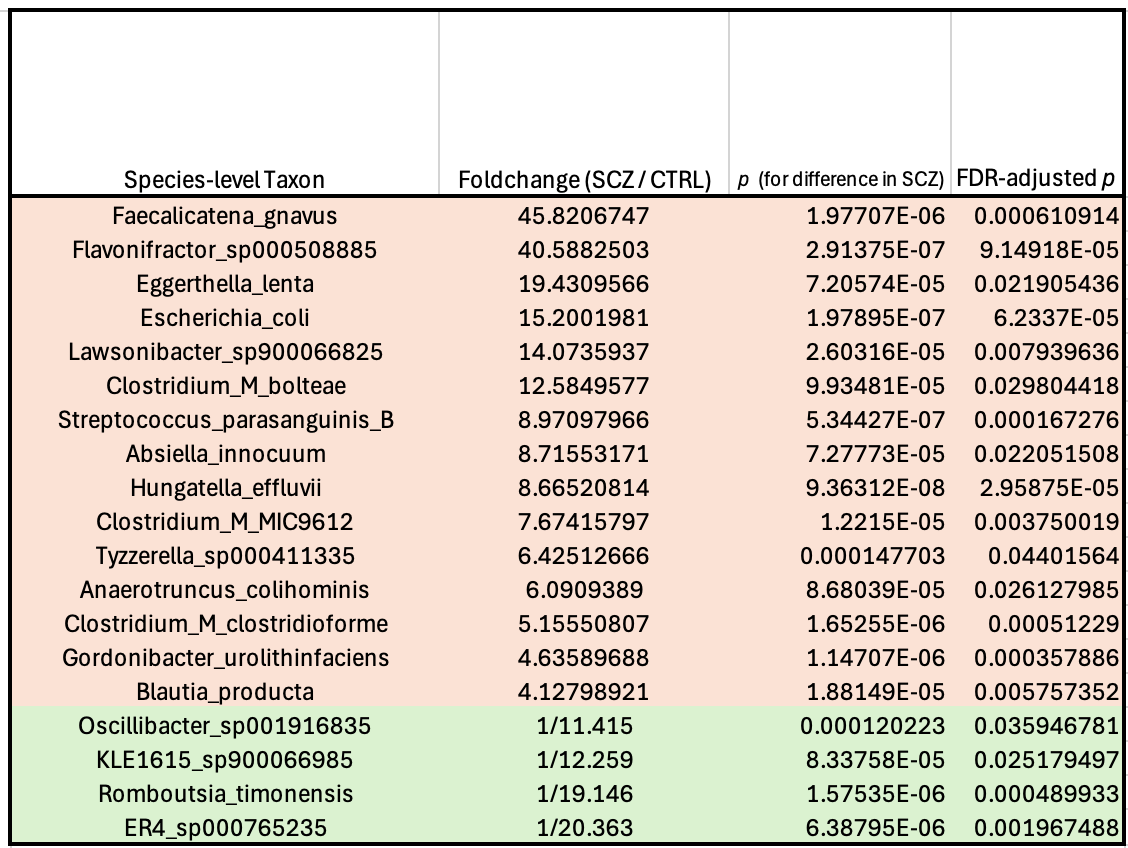
The authors concluded that this disrupted state was most likely a result of the patients’ medication, noting that the extent of the changes was fairly well correlated with the dose of antipsychotics they were on. I’m skeptical of this explanation for a couple of reasons, though: For one thing, patients who have more severe symptoms get prescribed higher doses of antipsychotics…so if certain bacteria cause the symptoms, you’d expect to see a correlation between drug dose and the abundance of those bugs. For another, there’s at least one study of “drug-naïve” schizophrenics which reports the same microbiome pattern in patients who had never taken antipsychotics. That study also found that six months of treatment with an antipsychotic (risperidone) decreased levels of these bacteria.1
But the thing that initially made me go “wait a sec” is that I happened to recognize a lot of the names on that list, from my days working at Holobiome—a microbiome science startup back in Boston, where we were investigating the gut-brain axis. Faecalicatena gnavus, the Clostridium_Ms, Absiella innocuum—all of these bacteria were on our radar because they’re known to possess an aromatic amino acid decarboxylase (AADC) enzyme—giving them the ability to turn the amino acids tryptophan and phenylalanine into tryptamine and phenethylamine, respectively.
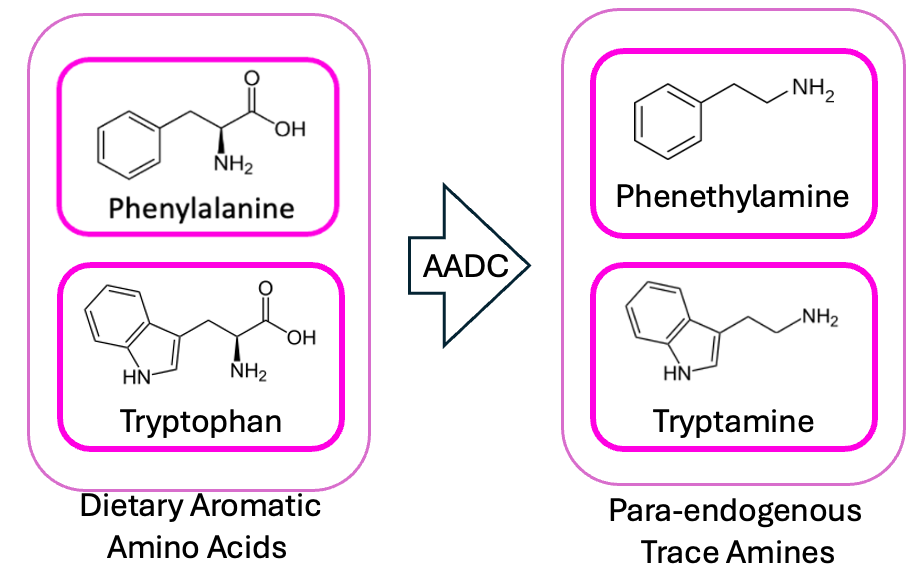
And pharmacologically, this is an interesting fit for some of the symptoms of schizophrenia: tryptamine is the core structure of most of the classic psychedelics (e.g. psilocybin), and phenethylamine acts a lot like amphetamine in the brain. This jives with what we know about the biochemistry of psychosis, in that—given enough amphetamine—an otherwise perfectly sane person will start rambling about vast and ancient conspiracies, trying to pick nonexistent bugs out from under their skin, plotting to annex Austria etc.
Digging into this possibility, I found support in the supplementals of a meta-analysis of metabolomics/metagenomics studies, which showed that—in multiple independent cohorts from around the world—fecal abundance of many of these schizophrenia-associated bacteria is tightly correlated with the amount of tryptamine and phenethylamine in the bloodstream.
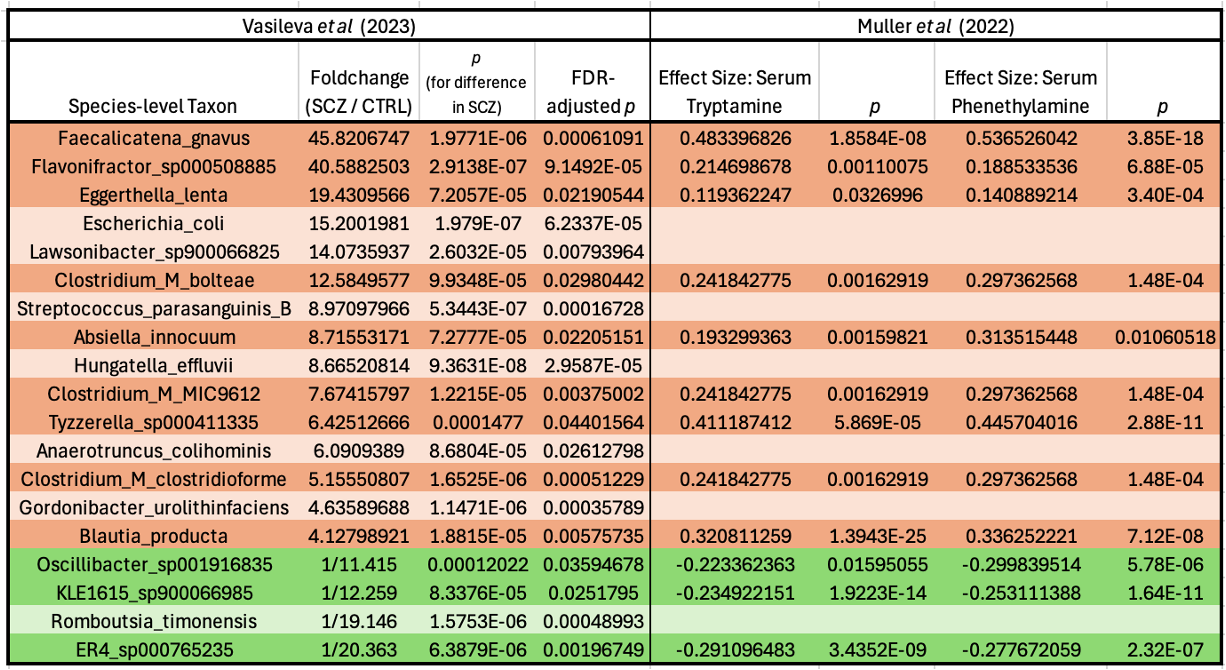
The ones that are elevated in schizophrenia are positively correlated with levels of these psychoactives, and the ones that are depleted in the disease are negatively correlated. Neat!2
But I could tell there was more to it: a few puzzle pieces that had yet to fall into place. I speculated a bit at the end of the original post, about what those might be—but nothing felt solid enough to really stand on.
Now it’s time for the rest of the story.3
The Pharmacodynamics Problem
My main point of dissatisfaction with the original theory was that, although phenethylamine and tryptamine act a lot like known psychoactive drugs in the brain, they only do so for a very short time. The enzyme monoamine oxidase (MAO) breaks them down very quickly, meaning phenethylamine’s half-life in the bloodstream is only about 5 minutes, and tryptamine’s is even shorter.
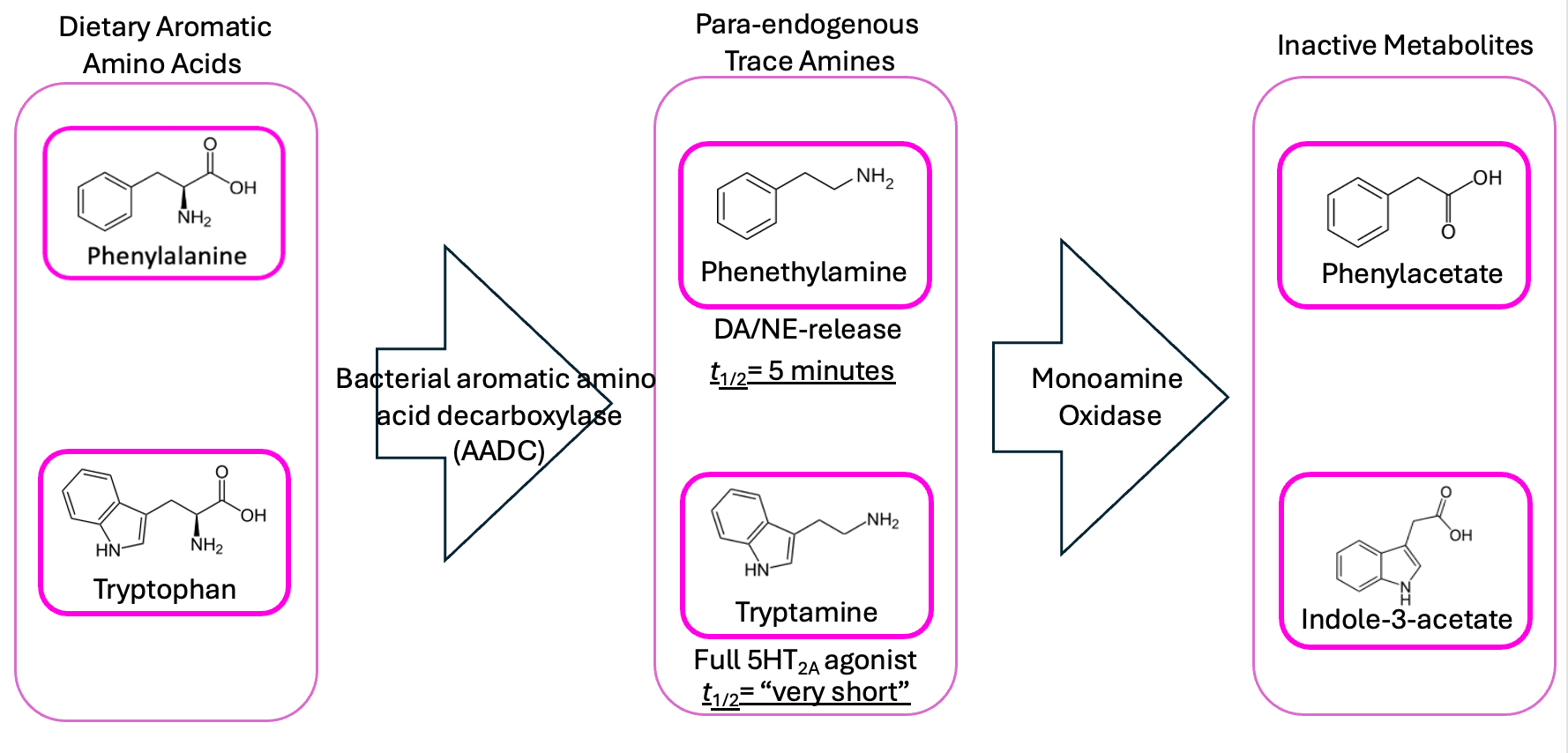
So putting someone on an in-house phenethylamine/tryptamine slow-drip shouldn’t do much (or at least not for very long), unless the doses were massive—unreasonably high, compared to the amount of tryptophan and phenylalanine in our diets.
My first try at patching the hypothesis invoked N-acyl amides, compounds that have been reported to reduce production of MAO, and which are correlated with some of the same bugs found in schizophrenia. But I didn’t like this, partly because the evidence around N-acyl amide production by gut bacteria isn’t very solid. Those same chemicals are used in plastics manufacturing and in industrial agriculture, so they might owe their presence in blood to something like processed food consumption—or even to the tubes that the blood is collected in.4
So it was a few weeks after I published the second schizo-post, with this pharmacodynamics problem rattling around in my head, that I went back to the supplementals of the Vasileva et al paper and started browsing. One of the tables in their datafile was a metabolic pathway analysis, which I hadn’t looked too closely at the first time—I was too gobsmacked by the differences in species abundance to pay much attention to anything else.
But going beyond taxonomy (i.e. describing things in terms of their genus and species) is often critical in microbiology, because there’s an astounding amount of “give” in the definition of a bacterial species relative to the kind of organisms we’re used to at the macroscopic scale. Thanks in large part to their ability to pick up genes from the environment wholesale and put them to use, two strains of E. coli can have as little of their genome in common as a partridge shares with a pear tree, and this is where it can be helpful to say “forget about what it’s called, tell me what it can do” and directly compare the abundances of individual genes, or the pathways they comprise.
And looking at which gene pathways are differentially abundant in schizophrenics vs. healthy controls reveals a surprising signal—one that finally made the rogue puzzle piece quit its rattling and snap cleanly into place.
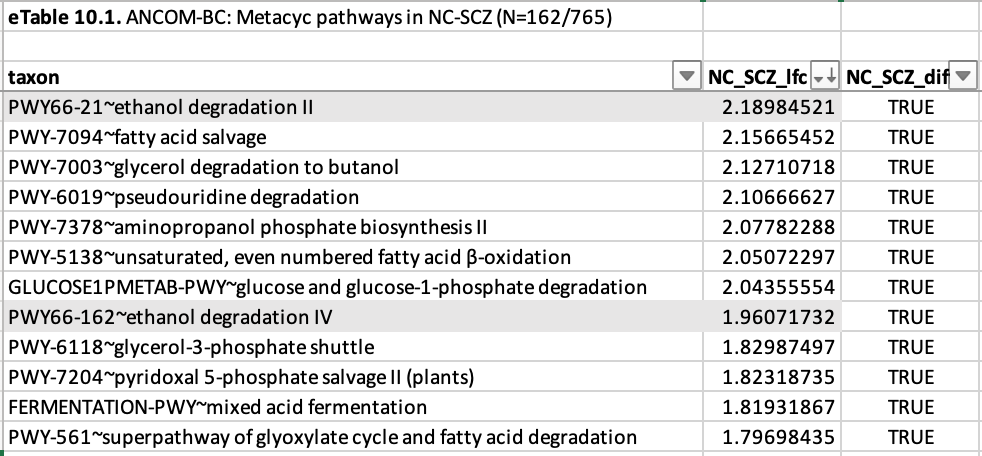
“Ethanol degradation”—two different kinds!
Now, you’re probably thinking: “So what? schizophrenics probably drink more.” But I don’t think that’s what’s going on here, for a couple of reasons.
For one thing: they told us so. Self-reported alcohol use was actually lower among schizophrenic patients than healthy controls. And yes, patients lie to doctors all the time, but consider that this study was done in Australia, where I imagine it’s actually quite difficult to keep pace with the “cognitively normal” controls, in terms of alcohol consumption, if you’re playing with the handicap of a serious mental illness. What’s more: alcohol is absorbed very efficiently in the stomach and small intestine, and metabolized in the liver—so while heavy drinking can change the composition of the microbiome, studies have shown that it does so by flooding the system with acetic acid—the end product of alcohol metabolism.5 You wouldn’t expect it to lead to an increase in ethanol degradation genes.
So if there’s any substantial amount of alcohol degradation happening in the microbiome, that means alcohol production must be happening there as well. And while we typically think of alcohol production as a feature of yeasts like Saccharomyces, the capacity is also pretty widespread among bacteria, including some of the bugs you find in the human gut. And in particular:

Now, if you’ve been following along through Parts I and II of this three-part Christmas special, you’re pretty well prepared to understand where this goes next.
See, when alcohol gets broken down for energy—whether by your liver or your gut bacteria—it first gets turned into the toxic acetaldehyde, then into the harmless acetate. It’s in that between-time when pretty much all the unpleasantness associated with drinking, from the carcinogenicity to the hangovers, arises.
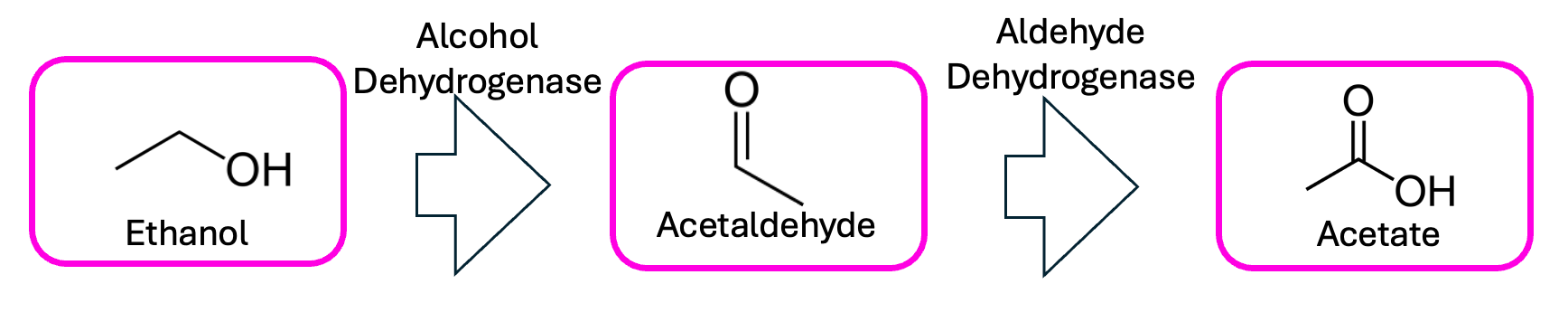
This is why people of East Asian descent typically have a harder time with booze; they have a less-efficient version of the enzyme that performs that second step, meaning every molecule of alcohol spends longer wreaking intracellular havoc as acetaldehyde. This is also how disulfiram—one of the most effective drugs to treat alcohol use disorder—works: inhibiting that second step turns even a single drink into a recipe for an instant headache and hangover.
But as we discussed in the last couple of posts, when you have an aldehyde like acetaldehyde and an amine (like tryptamine or phenethylamine) in the same solution, the aldehyde can spontaneously stick to the amine and cause it to cyclize. In the case of tryptamine, the thing that results is a beta-carboline. With phenethylamine, you get an isoquinoline.
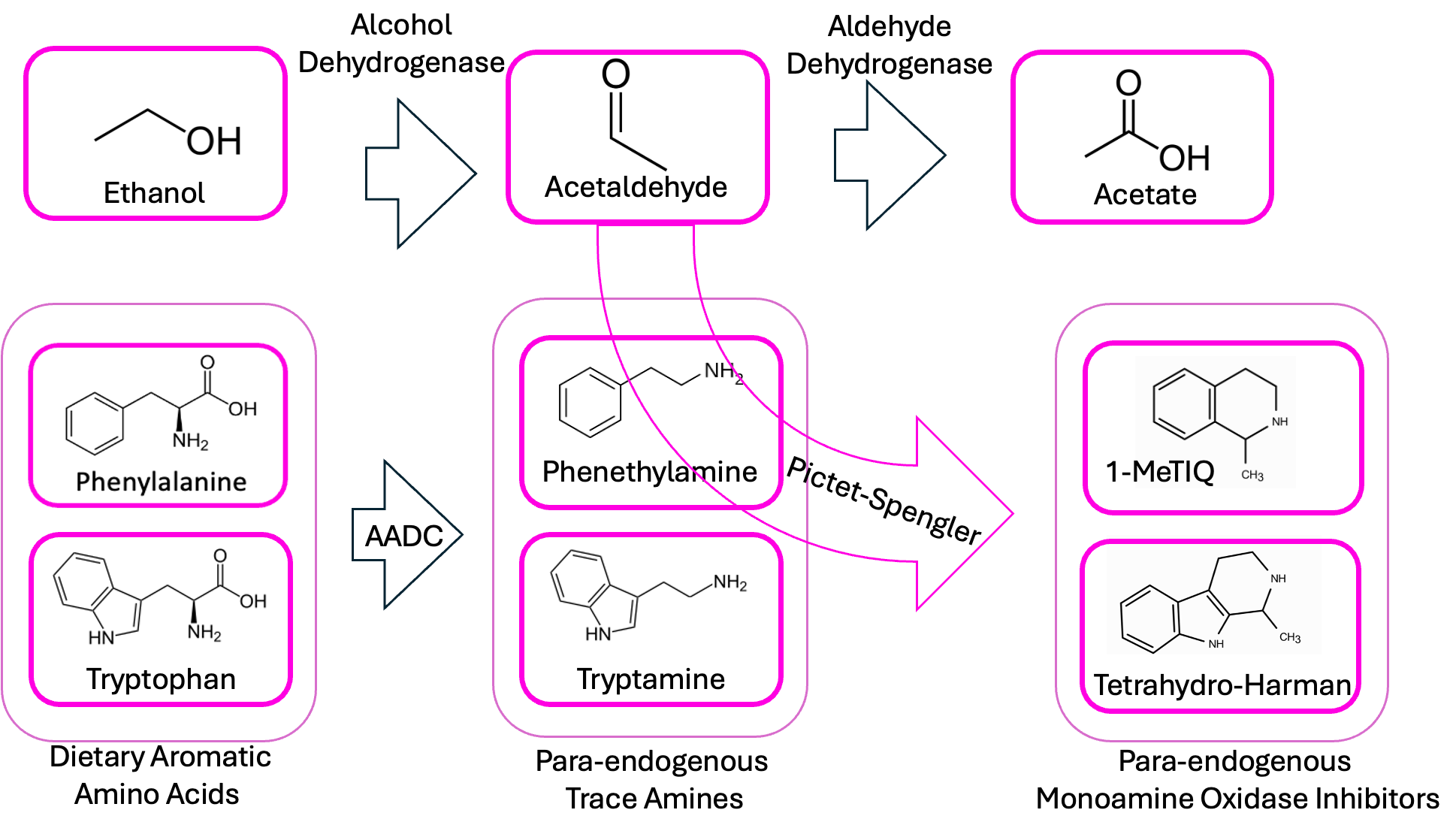
And this is amazing because beta-carbolines and isoquinolines are known to act as MAOIs. So think about this in the context of ayahuasca, the South American psychedelic tea (of “one-shotting turbonormies” fame). The active ingredient in it is DMT—dimethyltryptamine—but DMT isn’t orally active on its own, because MAO in the liver destroys the drug before it gets into circulation. That’s why the essential second component of ayahuasca is an herb that contains a natural beta-carboline, to run interference with your liver and give the DMT a chance to get to the brain.
As a result, a bacterium which produces both tryptamine/phenethylamine and alcohol (and thus aldehydes, and thus beta-carbolines) seems like a two-in-one recipe for psychosis, giving you both the psychoactive main ingredients and the adjuvant that makes them long-acting enough to cause problems.
This would also explain why the disease can’t necessarily be pinned to one bug: imagine Blautia producta has the AADC enzyme, but not the ethanol-production pathway, and Streptococcus parasanguinis produces substantial amounts of alcohol, but doesn’t have the AADC enzyme. If you had high Streptococcus alone, maybe you’d get Fatty Liver. If you had high Blautia alone, maybe you’d have something like depression or ADHD, since elevated levels of phenethylamine are thought to cause the brain to produce more MAO—deactivating neurotransmitters like serotonin and dopamine in the process. If you had both, then you’d have a recipe for the Bad Soup.
So this is another manifestation of “the problem with beta-carbolines”. It’s not that nobody ever imagined that they might be involved in psychiatric disease; it’s that—as discussed in the first post of this series—most of the work to investigate this possibility was done before we discovered that they can form spontaneously under conditions routinely used in chemical analysis. And when that unfortunate fact was found out, the neuroscientists collectively breathed a heavy sigh and basically went home, leaving the hypothesis for dead. But has anyone gone back and checked, using the proper methods discussed in that first post, whether schizophrenics have more beta-carbolines in their bloodstream than healthy controls?
The answer, as far as I can tell, is no.
Predictions
Now, it’s already known that schizophrenia—particularly the paranoid subtype—is associated with higher-than-normal levels of phenethylamine, and this aligns nicely with the inferences we can make by combining Vasileva et al’s microbiome data and the metagenomics/metabolomics dataset: if schizophrenics tend to have 45x the normal amount of Ruminococcus gnavus, and fecal Ruminococcus gnavus levels are tightly correlated with bloodstream phenethylamine levels, it follows that schizophrenia would be associated with high bloodstream phenethylamine levels.
This is why I subtitled this post “Some armchair-testable predictions about psychotic disorders”: one of the best ways to explore a new hypothesis is to see if the predictions it makes are consistent with the data that’s already available to us. So what else would this theory imply?
Well, let’s plot this all out, just to keep tabs on the major players here.
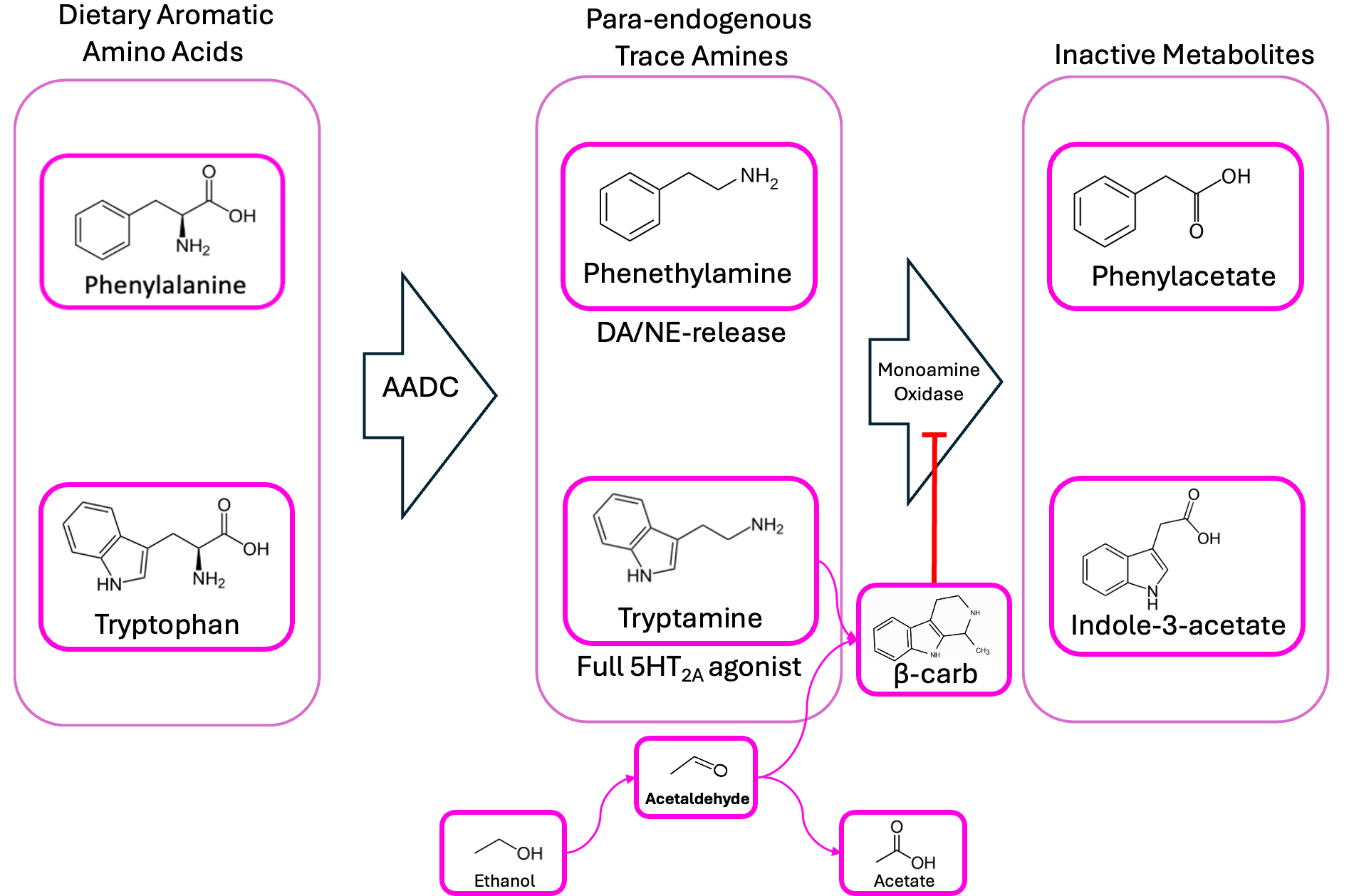
We’re proposing that schizophrenic patients’ gut bacteria produce too much phenethylamine and tryptamine, and that they’ve also got some microbiome-derived beta-carbolines/isoquinolines floating around, which prevent MAO from breaking down phenethylamine into phenylacetate. So, in addition to high phenethylamine, we’d expect schizophrenics also to have lower-than-average levels of phenylacetate, right?
And interestingly enough:
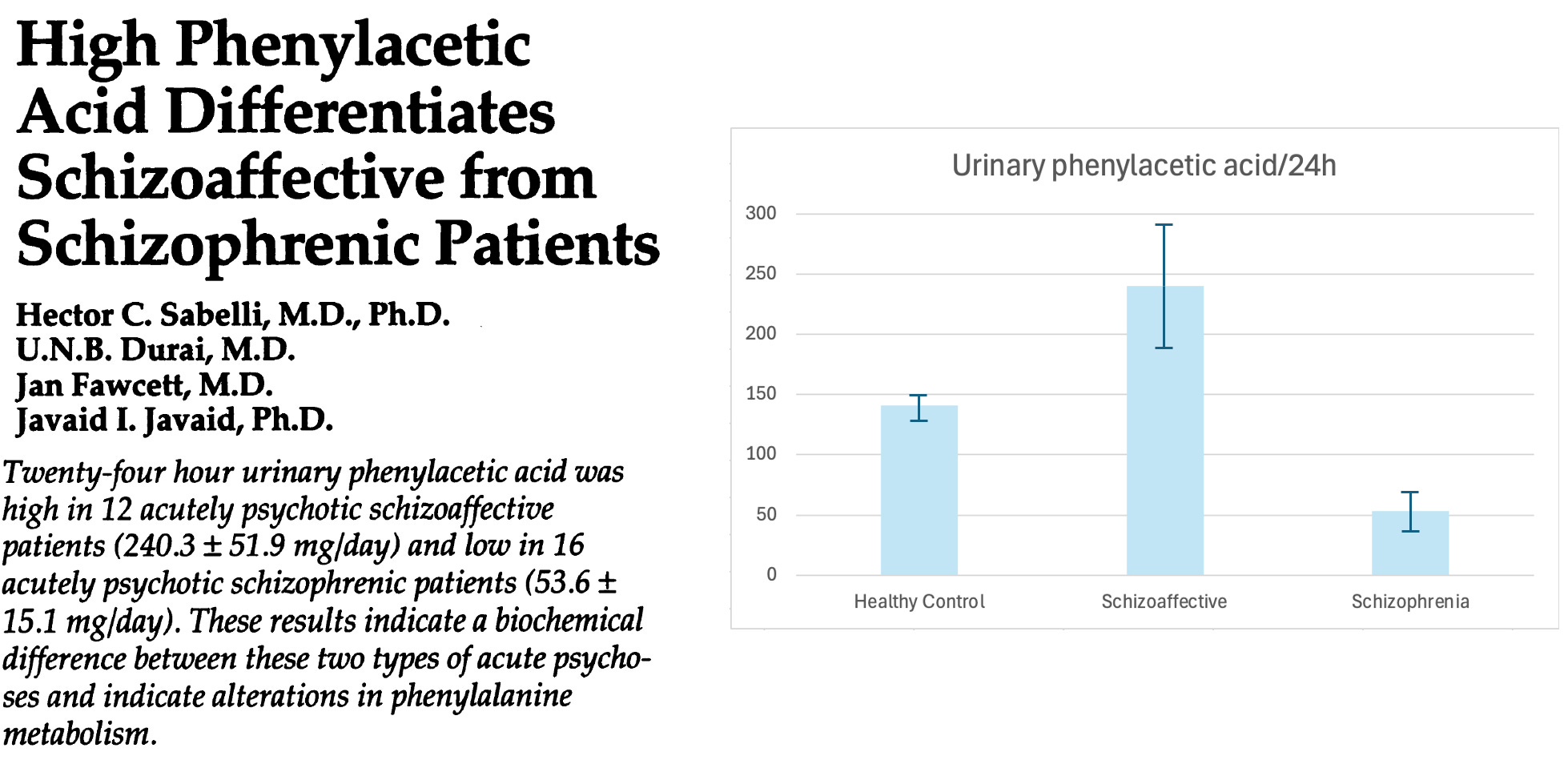
Schizophrenia is associated with low phenylacetate—less than half normal! But the paper I took the above clipping from also looked at patients with schizoaffective disorders, and found that they had high urinary levels of phenylacetate. What do we make of this?
Well, “schizoaffective” refers to patients who have symptoms of both mood disorders and schizophrenia. Bipolar is a good example: patients in a manic phase often spiral into a psychosis that’s clinically indistinguishable from that of schizophrenia.
This suggests a model of two diseases: maybe both schizoaffective and schizophrenic patients both have too much phenethylamine production happening in their guts, but only the schizophrenics have a substantial amount of aldehyde—resulting in the MAOIs that prevent these compounds from being broken down.
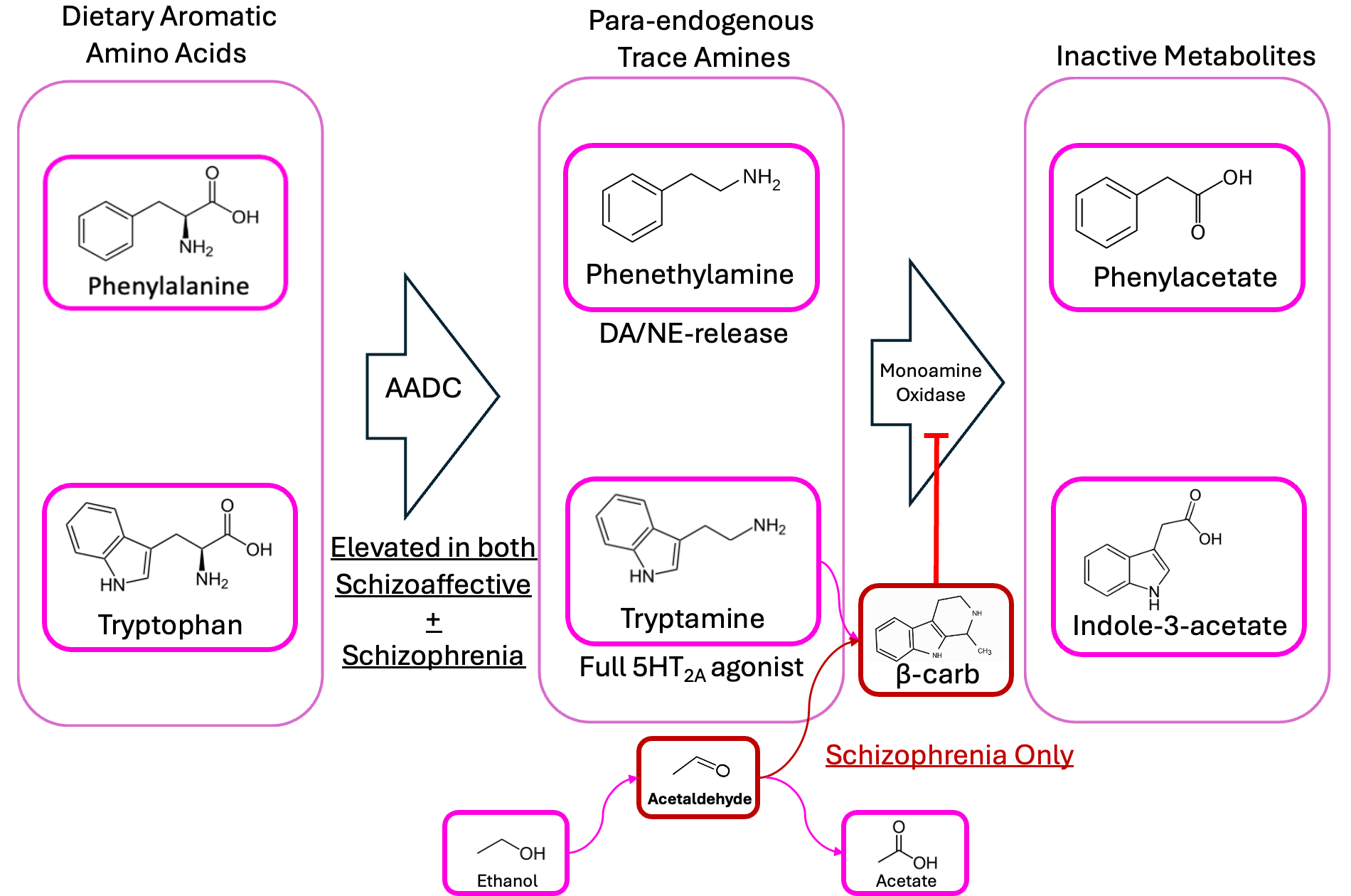
This naturally implies a heavy role for diet, as well: if you had a lot of a “two-in-one” bug like Enterocloster bolteae or Ruminococcus gnavus in your gut, but ate a strict carnivore diet—meaning no carbohydrates to ferment into alcohol/aldehyde—you might expect it to do wonders for your mental health.
And these observational tests are cute, but still pretty circumstantial. If you want to really put a hypothesis to the test, you’ve gotta get interventional.
So imagine we were feeling evil. Because in this model, psychotic disorders are just the high tail-end of the abundance distribution of a couple of common gut bacteria. That means there’s some subset of the population who only have one or two of the multiple requisite “hits” working against them—or maybe they’ve got all the necessary ones, but not that bad—only 10x average abundance of E. bolteae or R. gnavus. And so they’re struggling with some run-of-the-mill mental health issues, but they’re getting along more-or-less fine. How could we tip them over into not-fine territory?
Well, theoretically anything that increases the amount of acetaldehyde in the gut should encourage the formation of more beta-carbolines, right? And as luck would have it, there are drugs that do just that! We just learned about one earlier: Disulfiram, AKA Antabuse—the pill that takes all the fun out of alcoholism.
So this is our next armchair-testable prediction: If our model were correct, and there’s endogenous alcohol production and degradation happening in the gut, it shouldn’t matter whether patients are drinking: we’d expect psychosis to be an occasional side-effect of disulfiram.
And as it turns out…

This paper from 1967 reports acute psychosis as a side effect of disulfiram in between 2% and 20% of patients taking disulfiram!
Now, this is potentially a point in favor of the model, but it isn’t as clean-cut of evidence as I’d like. It turns out that, because one of disulfiram’s metabolites chelates copper, the drug can also inhibits dopamine beta-hydroxylase, the enzyme that converts dopamine into norepinephrine. Findings around this are somewhat inconsistent, but there’s a world where elevated dopamine levels resulting from DBH inhibition contributes to this phenomenon more than spontaneous MAOI formation. To suss out the real driver, you’d have to look for beta-carbolines and similar compounds in the bloodstream of someone in disulfiram psychosis.
So let’s look a little upstream. The crux of the hypothesis is that bacterial AADC in the gut is responsible for the disease. If that’s the root of where things go wrong, the ultimate testable prediction is that giving someone a drug which inhibits bacterial AADC enzymes should bring a person out of psychosis and keep ‘em that way. Of course, you’d need to find a drug that only hits the gut-bacterial version, because the same kind of enzyme is what lets your brain convert L-DOPA into dopamine and 5-HTP into serotonin.
But as luck would have it, there’s already an FDA-approved drug that works by inhibiting AADC. What’s more, it doesn’t cross the blood-brain barrier—meaning it should interfere with bacterial AADC in the gut, without inhibiting neurotransmitter synthesis in the brain.
It’s called carbidopa.
Carbidopa’s main use is as an add-on in Parkinson’s disease, a neurodegenerative disorder involving a breakdown in the dopamine biosynthesis process. Because the neurons that let you control your muscles run on dopamine, deterioration of that signaling machinery results in the disease’s characteristic tremors. By giving someone L-DOPA, which is converted to dopamine by AADC, you can restore dopamine levels in those motor neurons enough to stave off the symptoms of the disease pretty effectively, at least for a while.
But if you just give someone L-DOPA by itself, a lot of it goes to waste—getting converted into dopamine in the gut and liver before it has a chance to get into the brain where it can help with motor-neuron signaling. And while L-DOPA crosses the blood-brain barrier, dopamine itself doesn’t; it gets scrubbed out by MAO. Hence: giving someone a “peripherally restricted” AADC inhibitor like carbidopa along with their L-DOPA ensures that more of the latter reaches the brain intact.
And this is why, if you bring up “carbidopa for schizophrenia” to anyone who works in neuropsychiatry, they’ll probably balk. At first glance, it’s a counterintuitive idea: carbidopa’s job is to increase dopamine levels in the brain, and schizophrenia is generally understood to be the result of too much dopaminergic activity. After all, the most effective antipsychotics are all dopamine antagonists—shouldn’t carbidopa make things worse?
But if our hypothesis about bacterial AADC is correct—and provided that the AADC enzymes of various gut bacteria are similar enough to the human version to be susceptible to carbidopa’s inhibitory effects—you’d expect carbidopa to hit the disease basically at its root cause. No bacterial AADC, no tryptamine and phenethylamine, no MAOIs. But the problem with “armchair” hypothesis-testing is that it requires someone to have actually tried the thing—and because this sounds, naïvely, like a terrible idea that would make patients worse, I wasn’t particularly optimistic when I went looking in the literature for case reports.
But lo and behold, there is one.

Back in 1984, some research-clinicians at the NIH were lucky enough to have a patient with both a psychotic disorder and a history of stimulant abuse—and they were wise enough to listen to her when, feeling a break coming on, she told them: “Guys, I’ve taken a lot of speed in my day, and whatever’s going on here feels a lot like speed”. Understanding the similarity between amphetamine and phenethylamine, knowing that she had elevated urinary concentrations of phenethylamine, and reasoning that this must come from decarboxylation of phenylalanine by AADC, they tried carbidopa.
It worked. After 7 months free of psychosis, they did a double-blind withdrawal test; she became delusional again, almost immediately—and then recovered almost as soon as the drug was reintroduced.
It would be another thirty years before Fischbach et al reported that some common human gut bacteria possess an AADC enzyme that’s functionally similar to the human version, giving us the theoretical backing to understand why this worked—and why it might work in other patients just as well as it did in her. But knowing what we know now, and especially in light of the fact that the key signature of the schizophrenia microbiome is an overabundance of phenethylamine-producing bacteria, it seems like it deserves a more thorough exploration.
It’s not fully in the wheelhouse of my vision for Constellation, but I’m filing a patent around this one—something like “Use of inhibitors of bacterial aromatic amino acid decarboxylase for the treatment of psychiatric and mood disorders, particularly those with psychotic features”. Finding a new use for an approved drug is a biopharma investor’s wet dream, because 90% of the cost and failure-risk of bringing a new drug to market is in the Phase I and Phase II studies…things that are already taken care of for an existing drug like carbidopa or its Canadian cousin benserazide.6 If we can get a new treatment out there that addresses these disorders in a more fundamental way than existing drugs—and for a fraction of the cost and effort of bringing a new chemical entity to market—I feel like we’ve practically a moral obligation to make that happen.
More importantly, though, if we’re willing to get out of the armchair, this is information that any of us can use now. In the US, doctors have enormous leeway in the form of “off-label” scrips: any approved drug can be prescribed for any condition, as long as the prescriber has some rationale to think it’ll help. They already sell carbidopa as a standalone drug (i.e. without L-DOPA), under the trade name Lodosyn. If you or a loved one are struggling with a psychotic disorder and your psychiatrist tends to be open to experimental approaches, you can bring them the case report—hell, send them this post—and ask if they’ll let you try carbidopa. If they do, drop me a line and let me know how it goes.7
I’ve already devoted a lot of words to this topic, and spending too long with an idea in theory-space without bringing it out into the real world through experiment invariably starts to turn you into this guy—
…so for now I’ll just say thanks for reading—and watch this space. You’ll hear more on this one when I have data.
—🖖🏼💩
Shoutout to Scott Alexander of ACX for digging this one up! As iron sharpens iron, hey?
I should note that this is not obvious at first glance, because bacterial taxonomy is a mess. The genus known as “Clostridium_M” in Vasileva’s supplementals is known as “Enterocloster” in the metabolomics meta-analysis dataset, so if you want to compare between the two you have to use GTDB’s Taxon History tables to look up the equivalences. Or you can just really know your shit.
Famous (not-)last words.
In this latter case, you could imagine the microbiome’s influence on lipoprotein (LDL, HDL) characteristics mediating a correlation between the relative abundances of certain bacteria and the leach rate of these amphiphilic “slip agents”.
Certain gut bacteria, particularly Anaerobutyricum hallii and Anaerostipes hadrus, can consume lactic and acetic acids to produce butyrate—a very salutary metabolite which you otherwise only get from the fermentation of dietary fiber. Since Bifidobacterium, Lactobacillus, etc. convert lactose into lactic acid, and your liver converts alcohol into acetic acid, I am forced to conclude that cheese and beer, consumed together, count as a vegetable. Strangely enough, this is not the first time we’ve come to this conclusion on the blog, but through an entirely different rationale. Parallel lines of evidence!
Lately I’ve been seeing TV ads for a new antidepressant called “Auvelity”, which—when you read the fine print—is just Wellbutrin dipped in Robitussin.
I hope it goes without saying: If you’re a clinician reading this and you might be interested in spinning up an investigator-initiated trial, please reach out to SDSkolnick@gmail.com. I know a lab that can do the analytical chemistry.
If this story is true, it seems like we could also intervene by increasing aldehyde dehydrogenase activity? Zbiotics at least claims to do this.
I have a friend who had been experiencing slight auditory hallucinations, constant music, for his entire life. Doesn't report any all-out psychotic experiences, but does subjectively seem kinda wonky, with weird opinions and confusing lines of thought. He had helped me with some n=1 psychiatric experiments I wanted to run in the past, so I'm thinking, if you need someone of this profile to perform some tests on themselves, I can ask him.