

A former coworker hit me up this week. Her mother has ALS. It’s bulbar; the most aggressive kind.
“She’s in free fall”, my old friend says. Still all-there, mentally, but her motor neurons are flickering out, one after another. Months, if not weeks, ‘til the ones controlling her lung function go. It’s heartbreaking. It’s happening so fast.
She wants to know if there’s a gut microbiome angle on the disease, if there might be something we can do to slow it down—just to buy them some time.
I get these asks every so often. Every time, I take it dead seriously. Maybe it’s a vote of confidence, from the people who really believe in me and the work I’m doing—that they trust me enough to ask my advice on their loved one’s health. Maybe it’s just desperation; banging on the door of the witch doctor’s shack, once the more respectable physicians have started trotting out phrases like make her comfortable, complete with carefully practiced brow wrinkles, mouths set just-so.
Either way, I always try. Even if I don’t know much about a particular disease, I’ll do what I can to learn about it—do a lit dive to see if there have been studies of the gut microbiome in patients. Half the time, getting up to speed on the biology of a disease like ALS isn’t hard, because we still don’t know shit about fuck about what’s actually driving the disorder, in spite of decades of research.
Everyone wants to know if there’s anything we can do.
There usually is.
The Process: A Guided Tour
My first check is usually a literature search for any reports around fecal microbiota transplant (FMT) in a given disease, because it’s the most general test of microbiome involvement—whether by the presence of something nasty or the absence of something protective.1
A quick lit search turns up a small study—really just a pair of case reports—of FMT in ALS. Hey, I’ll take it. Both patients were hospitalized, trach’d at admission and dependent on mechanical ventilation to keep them alive. After FMT, both managed to wean off the ventilators and breathe independently again, at least for a while. Both also regained the ability to eat without choking. That’s pretty miraculous; typically once the doctors start stabbing holes in your neck, it’s all downhill.
So this is great! If we can get my friend’s mom an FMT, we might be able to help her. But, for some deeply stupid reasons, that’s easier said than done—at least if you want it done by a doctor. But maybe we can learn a little more about the disease from this FMT study, see if there’s anything we can do in the interim to buy us some time. Fortunately, they sequenced both of the patients’ microbiomes, as well as the donor’s, before and after the treatment.
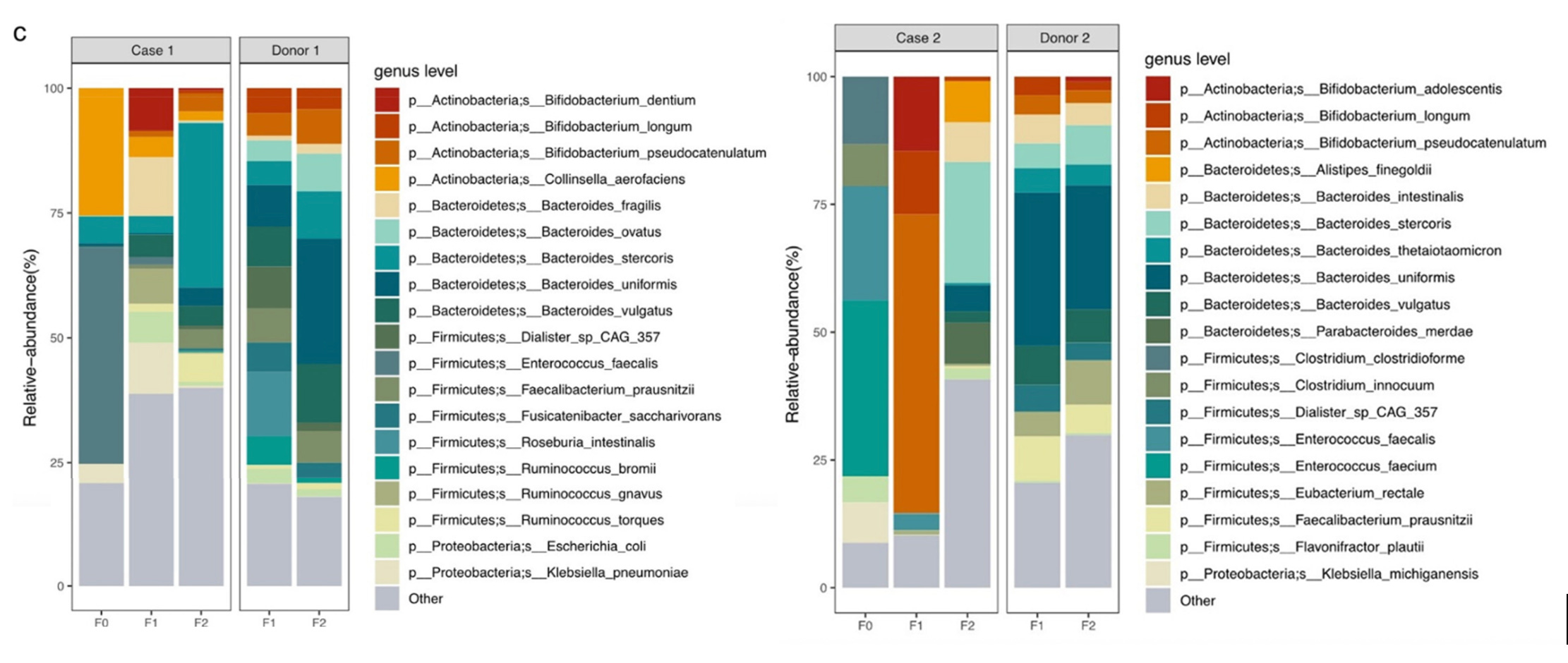
Now, the most obvious thing that’s out of whack here is the abundance of the genus Enterococcus. In both cases, it’s the dominant component of their microbiome at baseline; about 40% in Case 1, and well over 50% in Case 2. This is an absolutely bonkers amount of Enterococcus to have in your gut. In the general population, you might find Enterococcus in the guts of 1 in 25 people, where it might make up something like 1% of their microbiome.
The fact that it’s so drastically elevated in these cases—and that it’s suppressed to near-zero after the FMT, concurrent with improvement in the course of the disease—has my hackles up.2
So: what do we know about Enterococcus and its biology? I wrack my brain. An ICU nurse that I used to know talked a lot about VRE—vancomycin-resistant Enterococcus, one of the newer and deadlier “nosocomial” pathogens. A problem you only get in the wake of a course of antibiotics, when all the native flora are gone, so that all it takes is a few stray cells with the right resistance genes to waltz in and set up shop. That’s probably how these ALS patients ended up like this—you only get 50% abundance of one or two species if everything else was completely nuked at some point in the not-too-distant past. Of course, antibiotic use is associated with ALS risk.
I shake my head. A few more facts rattle out. It’s in the same family as Lactobacillus and Streptococcus, meaning it’s oxygen-tolerant—part of what helps it spread in settings like hospitals. There’s a veterinary probiotic that uses some strain of Enterococcus, although whether that’s E. faecium or E. faecalis escapes me. I survey this paltry collection of puzzle pieces; certainly nothing that looks like it connects to a paralytic motor neurodegenerative disease. But I know very little, and Google Scholar knows a lot. So we search: “Enterococcus paralysis”. Sort of a long shot—but it turns up something interesting. It's a study in nematodes, describing an experiment to figure out what makes Enterococcus a sometimes-pathogen.3
See, most of the common pathogens are pretty businesslike about it: Vibrio cholerae produces a protein we call cholera toxin, which causes salts to flood out of your intestinal cells and into your GI tract, which in turn makes you shit your guts out.4 E. coli produces lipopolysaccharides that piss off your immune system, and the nastier varieties make something called shiga toxin, a protein that prevents your cells from making new proteins, which triggers the cellular seppuku known as apoptosis.
But Enterococcus has no such protein toxins, as far as we know. It doesn’t produce LPS like E. coli does, or eat the host’s cells from the inside, like Salmonella. So how does it cause disease?

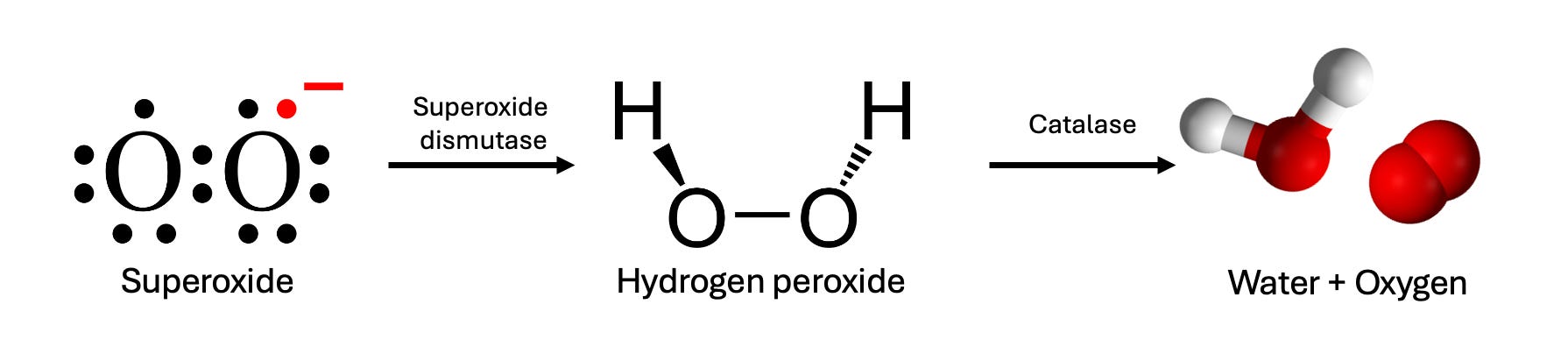
Apparently: by producing hydrogen peroxide—H2O2.
The chemical is a household name, thanks to its role as an all-purpose disinfectant and hair bleach, but it also plays an important role in our biology; it’s produced inside your own cells by an enzyme called superoxide dismutase, or SOD.
SOD’s job in your cells is to clear out a highly reactive form of oxygen that would otherwise wreak havoc; it does this by grabbing it and converting it into hydrogen peroxide. But hydrogen peroxide isn’t exactly safe either (as you might have guessed from the fact that it’ll bleach your hair), so your cells have another enzyme called catalase, which scrubs out the H2O2 as it’s produced, converting it back into H2O and regular O2
So the body is equipped to handle hydrogen peroxide, but only up to a point—and if fully half of your gut microbiome is an H2O2-producing species, you’re almost certainly well past that point. (Recall that, in my longform piece on phenethylamines and tryptamines in schizophrenia, we did some napkin math using the pretty safe assumption that there’s at least 200g of bacteria in the average gut microbiome…implying that both of our ALS patients had something like 100g of Enterococcus in their guts, pre-FMT.)
So here we have a rudimentary disease model: another simple twist on the classic concept of auto-brewery syndrome. Last time, we swapped out the yeast for Ruminococcus gnavus or a similar species, and the alcohol for phenethylamine and tryptamine. This time, it’s an Enterococcus, and our molecular monster-of-the-week would be hydrogen peroxide.
Nerve Bleach
Does this track with what we know about ALS?
Well, I don’t know shit about ALS, but I do another quick search: “Hydrogen peroxide ALS”, and…huh, would you look at that! A whole review on the interaction between the two.

For background: ALS is like a number of other mystery disorders, in that—while there’s no gene mutation that’s necessary for the disease to emerge—there are some that seem to be sufficient. Autism is one of these disorders: mutations to a gene called SHANK3 produce a syndrome that more-or-less fits the diagnostic criteria for autism, but this explains only about 1% of cases—the vast majority of people with autism have SHANK3 genes that are perfectly fine. Even so, the outward similarities between the SHANK3-mutation kind of autism and the other kind suggest that maybe we can learn something about the biology of the “other 99%” flavor of autism by studying what the SHANK3 gene does.
In ALS, likewise, something like 90-95% of cases are sporadic—meaning there’s no family history and no clear genetic cause. But the other 5-10% of the time, the disease is inherited, with patterns that tend to follow a particular gene variant—a mutant version of superoxide dismutase, the enzyme that produces H2O2.

Now, oftentimes when we talk about disease-causing mutations, we’re talking about loss-of-function mutations, where a random change to the genetic code alters a protein in a way that breaks it. Think of albinism, for example: typically a loss-of-function mutation in tyrosinase, the gene that makes melanin. And this is sort of the natural, default way to think of mutations, because life is a very finely tuned thing; if you open up the binary of a computer program and start flipping bits, you stand a good chance of breaking it, while the odds that you’re going to make it work better are pretty much slim to none.
But it happens in biology all the time…more often than the coding analogy would suggest, because—while a computer program is designed by the coder to fulfill a particular function in an efficient way—protein structures are arrived at more or less by the process of “open up the binary and start flipping bits”, meaning there’s often a lot of room for improvement. And that appears to be what’s going on with the mutant version of superoxide dismutase that appears to drive some cases of ALS: a gain-of-function mutation that makes it a more efficient producer of hydrogen peroxide than the standard-issue version.
So—because I’m grossly under-informed and ignorant of most of the research that’s been done in this field—this all seems pretty pat to me. If familial ALS can be caused by a mutant version of SOD1 that spins out too much H2O2, having a microbiome that does the same thing seems like a solid explanation for the sporadic version of ALS.
Now, like last time, this hypothesis is more about a molecular pathway than an individual species—just as Ruminococcus gnavus, Enterocloster asparagiforme, and certain species of Blautia can all produce tryptamine and phenethylamine, there are a number of other bugs that produce H2O2 under the same conditions as Enterococcus. Lactobacillus and Streptococcus are among the best-known, so you wouldn’t necessarily expect Enterococcus to be the only microbiome feature associated with ALS.
This isn’t as well-fleshed-out a hypothesis as it could be; but the fact that—fully ignorant of the link between ALS and the mutant SOD1 gene—I found my way to “These people ought to have way too much hydrogen peroxide floating around”? That’s good enough for me, for now—because it might be good enough for my friend and her mom. “Enterococcus infection” is something any hospital care team knows how to handle; we’ve just got to convince them to send a fecal sample to the bacteriology lab, see if there’s an unusual amount of Enterococcus or something similar, and—if there is—find an antibiotic that it’s susceptible to. That’s not a long-term solution, especially since overuse of antibiotics is how we get things like VRE…but maybe it can buy us some time.
—🖖🏼💩
At a high level, you can classify every kind of illness resulting from dysbiosis as either a disease of absence, or a disease of presence. If you've got high cholesterol because you're missing the microbes which are supposed to transform it into coprostanol, that's a disease of absence. If you've got multiple sclerosis because there's a colony of Clostridium perfringens in your gut churning out epsilon toxin, that's a disease of presence. In reality, things are never so cut and dry: because many microbes have antagonistic relationships with each other, an overabundance of one thing can be caused by the absence of something else, and vice versa.
In a forest, if a hunter kills all the foxes, you might get an excess of rats—an absence manifesting as a presence. Alternatively, if some rats just showed up one day and started eating all the nuts that fall to the forest floor, driving the squirrels out, you could have a presence manifesting as an absence. And, just to make things more difficult for us, maybe the rats showed up, edged out the squirrels, and the foxes starved because the rats aren't good to eat.
The complexity of these dynamics, compounded by the fact that the foxes, rats, squirrels, and everything else in this story are too small to see (and a quarter of them don't even have names yet) means that most of the time we're never going to get clear answers as to where exactly things went wrong, and our best hope of fixing things is to just burn the whole show down and pave over the ashes with an ecosystem that’s less fucked.
—excerpted from “Banger$ McGuffin’s DIY Guide to FMT, v1.7
I kind of hate color-coded relative abundance charts like this, because the legend always has at least twenty things on it, so you invariably end up having to open up a program like Paint to compare swatches and check whether that’s Bacteroides thetaiotaomicron teal or Enterococcus faecium teal. It’s Enterococcus teal.
The word “paralysis” is only used in passing in this paper, in reference to the effects of Pseudomonas aeruginosa in their worm model, so the fact that this paper turned out to be exactly what I was looking for is what—in a game of pool—would be called “slop”: you miss the shot you were trying for badly enough that you end up sinking a completely different ball. Bob Ross would be proud.
The actual mechanism by which cholera toxin does this is incredibly complex, Rube Goldberg-esque, and not interesting at all.
As usual from you, very fascinating and plausible-sounding hypothesis, though my expectations are tempered by the fact that in general most hypotheses don't end up panning out. Hoping for the best, though. Please let us know whether your predictions about her elevated Enterococcus (and about treating it) end up being right or wrong. As always, the best way to test these things are to see whether the predictions are accurate.
Loving this detective work you are doing. Is there any hint that smaller populations of microbes outside of the gut/mucosa might be having more direct impacts on various tissues and organs during disease states? Recent reports on the brain and tumour microbiomes has me wondering...