
The Sun
A Response to Astral Codex Ten's "Contra Skolnick on Schizophrenia Microbes"

So, first things first: A long time ago, my beautiful wife promised that when I broke 1,000 subscribers on my Substack, she would bake me a cake to celebrate. Well folks, I’m delighted to report that we’re now over 1300, and it was a delicious cake.

Now: in my last post I made some bold claims about the microbiome’s role in the etiology of schizophrenia. A few days later, prominent rationalist, psychiatrist, and Substack OG Scott Alexander happened to put up an ~8,000-word explainer on the problem of “missing heritability”, grappling with the fact that a lot of heritable traits like height, IQ, and—yes, mental illness—don’t map cleanly to the genome the way e.g. blue eyes does.
Between two or three people independently linking the piece in the comments of the “missing heritability” post, and me sharing it in his weekly “open thread” shortly thereafter, he finally decided to let us all know how he feels, putting out what one reader described as a “pretty damning takedown of [the] theory”.
I wouldn’t go quite that far, but he certainly didn’t mince words about disagreeing with me, and he gave his reasons why. And if there’s one thing I love about the rationalist worldview, it’s that it has no place for the notion of “agree to disagree”: If two perfectly rational people disagree, one of them is likely missing information the other has, or else is misinformed. And if, through argument, they can find the asymmetry in their knowledge-bases or worldviews, together they can get their facts straight, and whoever was wrong can change his stance.
In theory.
In practice, we're all human. Even a very rational person is susceptible to biases and fallacies like sunk cost. So when, over the course of your prolific career, you've dedicated an awful lot of words to exploring The Mystery of X—long explainers on why X is such a mystery, on its various aspects and manifestations, how we know it is not A, B, or any of the other letters down through W, and (just last week) “Eight Possible Explanations for the General Class of Phenomenon that X Seems to Belong To”…you get some inertia. And when some fresh-faced twat comes along and says “Oh, X? Not a mystery at all, that’s just well-understood-but-obscure thing Y”…it's hard to take him seriously, even if he's got a compelling argument.1
So the rebuttal is not unexpected. I might have preferred to explore these finer points in dialogue, rather than a format which feels a lot like skywriting…but I’m glad to at least see the guy’s objections outlined so I can address them, because they’re all eminently addressable.
As a warning: This is probably the longest substack post I’ve written, clocking in around 8k words. This is the inevitable fallout of making a post which proposes a grand unified theory of the biggest problem in the field, using fewer words than your average recipe blog SEO post. As noted at the top of the last piece, it was a transcript of a talk for which I was allotted a total of 10 minutes, delivered to an audience at a neuro-focused conference, so a certain amount of background was glossed over for the sake of brevity. I think the likes of Scott Alexander assumed that I had told as much of the story as there was to tell, and that he thus knew everything he needed to know in order to respond, when in fact I told the briefest possible version, because—unlike some people I could mention—I try not to make my readers slog through 8,000 words of gory details unless there’s a reason. But an idea this big necessarily takes a certain number of words to shore up and flesh out, so if you believed me about the five-minute version, you’re free to go if you like.
I. Discount of Monte Carlo
We’ll start off with Scott’s assertion that the following two statements are perfectly compatible:
-80% of variance in schizophrenia risk can be explained by the genome, and
-If your identical twin—who has the exact same genome as you—has schizophrenia, there is only a 30-40% chance that you will have schizophrenia.
(The term “concordance” will be used a lot below; this is what it refers to—when both members of a pair are the same in some way. Concordant=matchy, discordant=mismatch). Scott says:
[30-40%] is exactly the concordance rate you should expect from a polygenic condition where 80% of the variance is explained by genes.
That sounds wrong to me, but so did the Birthday Paradox thing until I saw the proofs on it, so I’m open to the notion that I don’t understand statistical variance, or that my intuition is the problem here. He goes on:
I discuss this example in Some Unintuitive Properties Of Polygenic Disorders and run a discount simulation that assumes an 80% genetic disorder with 1% prevalence and finds a concordance rate consistent with observed values. Here is a paper that does the full formal simulation and finds the same.
Okay but the thing is: No. You don’t. You fumble the one line of math involved, resulting in your simulation yielding a result within the (generous) target range you set for yourself. Then you get corrected on the math by a reader, implement the correction, find that the results no longer support your conclusion…and then you don’t change your conclusion, dismissing the discrepancy by gesturing at someone else’s work, which—according to commenters on your post—you have also misapprehended.


To break this down: Alexander is invoking this “simulation” as support for the plausibility of the claim that 80% of schizophrenia is genetic. The condition under which the simulation would support this claim is if it yields a twin concordance rate somewhere in the range of 0.15-0.5.
This is not a particularly tough mark to hit. Fully one-third of the entire range of possible outputs fall within it. You’d have pretty good luck just calling the “RAND()” function. But he still misses the broad side of the barn.
By way of explanation, he points out that 10% concordance is low out-of-range, so he’s not worried about it because any environmental similarities (same parents, born the same time of year) should increase it, and likely bring it up into the range of actual observed values. Obviously this is the point at which he should have said “hm, maybe clicking around in a spreadsheet full of randomly generated numbers is not a useful way to study a complex human disease.”
But now we find ourselves in a truly remarkable position, because according to Scott’s math: if 80% of schizophrenia variance is genetic, identical twins with no common environment should only have a concordance rate of 10%. So, say we go out in the real world and find ten schizophrenics who have identical twins. And we check the twins, and we find four of them also have schizophrenia—i.e. a concordance rate of 40%. Scott’s result implies that 3 of those 4 owe their matching disease status to things outside the genome.
So, for those keeping score at home: if the disease is 80% genetic, we’d expect 10% baseline concordance among identical twins, which is how we know that up to 75% of real-world twin concordance is due to things other than genes.
Does that sound right to you?
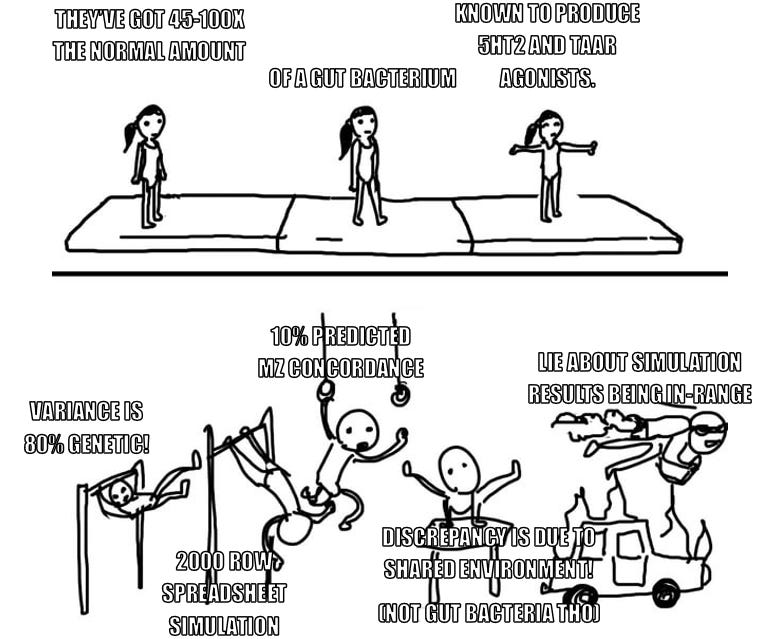
Maybe all this is technically possible under some weird definition of the words “risk”, or “genetic”, but if so, these definitions are so divorced from their commonsense meanings that it’s counterproductive to try and use them in conversation.2 I have to assume Scott isn’t just blatantly lying about the simulation result and hoping nobody clicks through, so either:
he forgot about the correction and has that whole situation wrapped in a bundle labeled “evidence schizophrenia is 80% genetic” in his mind, or
he used an LLM to write his rebuttal post, and this was sort of a “hallucination”.
The more I think about the latter possibility, the more likely it seems. It fits the pattern of confidently wrong assertion-with-citation that you get from current models, Scott just mentioned dealing with o3 hallucinations in the writing of a recent piece, and he seems like the kind of guy who would have a custom GPT trained on his corpus. I also thought “Contra Skolnick” came out suspiciously quickly after I posted in the Open Thread for the week, and figured he must have had it in the works since he got comments on the “Missing Heritability” piece…but that also fits with a timeline where he just had ChatGPT bang it out, looked it over once, and hit send. If this is the case, I will be absolutely furious, but also delighted that I got to live out a classic scifi trope.
So let’s move on, because all this is just the argument for why the genomic hypothesis shouldn’t be DQ’d from the get-go, on the basis of monozygotic twin concordance being below 50%. Centering the discussion on “Is the genomic explanation even mathematically possible, given what we know about genetics and statistics?” draws focus away from the real question at hand, which is: “How well does this hypotheses do at explaining what we see and what we know about this disease?”
So, just for fun: take the microbiome out of it. Imagine a miraculous sequencing breakthrough happens tomorrow, where they fix some long-overlooked flaw in the tech, and we suddenly find that—where most people’s genomes have only one copy of this aromatic amino acid decarboxylase gene—schizophrenics have, on average, 45 copies.
This would presumably be hailed by the Scott Alexanders of the world as rock-solid evidence that we’ve finally found the cause, or at least made a monumental breakthrough, because a 45x difference in a monoamine neurotransmitter-related enzyme blows every other gene correlated with schizophrenia right out of the fucking water, both in terms of signal strength and mechanistic plausibility. You’d still have to explain why it waits to kick in until a certain age, but we’ll get to that.
In terms of shedding light on the situation, the best the geneticists can do is five thousand mysterious SNPs, like stars in the night sky. There are a lot of them! And as they build better telescopes, they will discover more.
But they won’t make it any easier to see.
I offer you the sun.
There is just one. But it’s a good one.
II. A Loving Reminder to the Peanut Gallery
Scott’s “Contra Skolnick” opened by describing me as a “gut microbiome expert”, a label which I’ll wear with pride—but it sparked a few sniggering comments to the effect of “Mandy Rice-Davies Applies”, i.e. “Of course, as a gut microbiome scientist, he’s going to insist that schizophrenia is in the gut microbiome.” And while I mentioned this in the reply to those comments, I felt it worth reiterating here: I was not always a poop scientist. My undergrad was in physics, and I didn’t go to grad school; I had to hustle and muscle my way into this field, making risky moves and some real sacrifices, leaving a nice job as a science writer at APS for the chance to become…a turd-wrangler.
I chose to do so because, when the key facts about the microbiome—that it is at once both highly heritable and wildly variable—started to percolate into the public consciousness a decade ago, it became apparent to me that this could be as important for our understanding of biology as Mendelian inheritance was, and that it’s a promising potential explanation for many of the chronic illnesses that thus far have stumped our best and brightest.
This doesn’t mean I’m free of biases—if anything, I’ve got a lot more sunk-cost than someone who happened to be a microbiology major graduating in 2014. Worse, I have always been a relentless optimist, straining to see any way in which diseases like schizophrenia might not be hard-coded, but rather mutable, and therefore fixable. But I’m an empiricist, and I pride myself on that. I’m aware of my biases, and I go to great lengths to avoid letting my desire for something to be true impact my assessment of whether or not it is. Obesity is a great example of something where it would be nice, but I don't think we’re going to be able to treat it effectively with microbiome modifications like FMT, because a number of people have tried and failed.3 Point is: Experiment is king, and if getting rid of the tryptamine-producers in a schizophrenic person’s gut doesn't bring and keep them out of psychosis, then hey, I’ll happily take this hypothesis out behind the woodshed.
I would be quite surprised if it didn’t help, because—as I pointed out last post—a meta-analysis of metabolomics studies has found that the correlation between the abundance of Ruminococcus gnavus in the gut and the concentration of tryptamine and phenethylamine in the bloodstream is the strongest correlation between any microbiome feature and any serum metabolite, period. That is to say: if we know literally one thing about the gut-brain axis, it’s that having a lot of gnavus and Enterocloster in your gut microbiome means having a lot of tryptamine and phenethylamine in your bloodstream (and thus your brain)4 Maybe removing them wouldn’t do anything, because it turns out these compounds have nothing to do with the pathology of schizophrenia! But it would be a very weird coincidence.
So, apologies for the long digression here, but I wanted to directly address the notion that I have conveniently formulated some theories wherein my expertise places me at the center of the universe. I saw the center from afar, and I have moved to it: Anyone who knows me personally will tell you that I have been yammering on about how many of these problems look like nails, even back ten years ago when the nearest hammer was located across a river of shit, and up a high cliff (also of shit).
The other reason I bring up my time as a physicist is because one of the arguments in Scott's rebuttal—the first paragraph, below—reminds me of the racket that the particle theoreticians have going on.
3: Also, scientists have looked for schizophrenia genes, and can only find about 1-2% as many as they were expecting.
This is the “missing heritability” problem, common to all polygenic traits. See my Missing Heritability: Much More Than You Wanted To Know. The leading explanation is that our current genetic screening methods aren’t good enough to pick up rare variants; an alternative explanation proffered by some geneticists is that there are lots of invisible gene x environment interactions.
In either case, schizophrenia doesn’t have much more “missing heritability” than anything else, and it would be surprising if every single trait - from height to educational attainment - was determined by gut microbes.
The appeal to our technological limitations is a powerful one: the supersymmetry guys and string theorists love to propose a grand unified theory, an ultimate explanation for all that is…which conveniently can’t be tested without a particle accelerator the size of Moldova, and which would cost the entire GDP of Moldova to build. So, tenure secured on the strength of a prestigious and effectively unfalsifiable theory, the profs can get back to theorizing about the existence of glueballs5 and teaching the occasional cosmology class, to ensure that the next generation of drone engineers is a well-rounded bunch. On the off chance that some government or coalition comes along that’s crazy enough to actually BUILD the damn thing, they can look forward to consulting on the construction, but rest easy knowing they'll be well into retirement by the time the beamline is operational and their ideas risk being put to the test.
All this to say: I would love to see the day where a new breakthrough in sequencing tech turns out to be biology’s Great Moldovan Hypercollider, but I'm not holding my breath, because this is the same spiel we've been hearing since the earliest days of gene sequencing.
On the second paragraph in the pull quote above:
I agree, it would be surprising if everything from height to educational attainment were determined by gut microbes. I don’t consider that very likely. But the unlikelihood of every such trait being gut-bacterial has no bearing on the likelihood that any one such trait should be.
Laid out in formal logic, the argument Scott seems to be making is:
Given: schizophrenia is a typical part of the set of “traits with substantial missing heritability”.
If: Schizophrenia is gut-bacterial in origin
Then: The rest of that set, e.g. IQ and height, would also be gut-bacterial in origin.
Given: it’s highly unlikely for IQ, height, etc. to be gut-bacterial in origin.
Therefore: Schizophrenia is unlikely to be gut-bacterial in origin.
Laid out like that, hopefully it’s clear that the if-then proposition involved is a classic squares-and-rectangles misunderstanding. This is a weirdly basic fallacy that I wouldn’t expect from a central figure in a movement that prides itself on logical acumen. Maybe more support for P(o3), or just a sign that Scott’s realized he can deploy long explainer articles like smoke bombs, and that it doesn’t really matter what he says after that, as long as it sounds punchy.
III. What Schizophrenia Does and Does Not Resemble
One of the main problems facing the field is that, if schizophrenia is mostly genetic, we need to be able to explain why mutations that are present in the genome from birth only start causing trouble once a person is basically fully grown. This is why phrases like “gene-environment interactions” and “two-hit model” get a lot of play in discussions of how the pathology of the disease might work. As Scott puts it:

And I’m glad he asked that question, because it’s one of the greatest strengths of a microbiome-centric explanation, not only for schizophrenia, but for practically any chronic and noncommunicable disease where a person is fine for most of their life, and then it seems like a switch just “flips” one day, and now it can’t be un-flipped.
Because, while your microbiome ordinarily exists in a pretty stable state of equilibrium, it’s still the most labile component of your biology. The relative abundances of various bugs wax and wane from day to day, from poop to poop—depending particularly on what kind of meal those poops started out as. But provided you don’t swing from full veganism to a carnivore diet overnight, all the species that are there one day will likely be there a week later, at roughly the same proportions.
But say you get norovirus, and spend three days straight shitting your guts out—or you get Lyme disease, and have to spend a month on some very heavy-duty antibiotics. This is the kind of ecological upheaval that can tip the ecosystem into a new equilibrium: just as stable as the previous one, perhaps even more stable, but not for the better. Maybe your Phascolarctobacterium has gone extinct, and then suddenly you’ve got three times as much Dialister, because those are basically the only two genera that eat can eat succinic acid, and your Bacteroides produce a lot of that. And then, over the course of the following month, you suddenly come down with a mysterious disease that has a stupid fucking name like “ankylosing spondylitis”, and which—for some strange reason—comes with a free side of IBS.
This is where the Chronic Lymers and the Branch-COVIDians come from: You get sick for a week and get over it, but you’re never the same after. In the case of norovirus (and maybe even COVID) this is sort of a consequence of the disease itself, but with Lyme it’s more the collateral damage from the treatment: burning down the jungle that is your gut ecosystem. Things will grow back, but not quite the same as before. And if suddenly you find yourself with too much bamboo, good luck, because burning it all down again is probably just going to get you even more bamboo.
So, in light of this, think about what we know about Ruminococcus gnavus’s prevalence and abundance in the human gut. It’s detectable in about 50% of people’s fecal samples globally. (Notably: those people have 33x the odds ratio of cardiovascular disease vs. gnavus-negative, if I’m reading this study right [I can’t be reading that right; someone check me on that?])
So it’s prevalent: a lot of people have it. But how abundant is it? How much do they have?
For the most part, not a lot; it’s typically less than 1% of the bacteria in your gut—and usually more like 0.1% or 0.3%
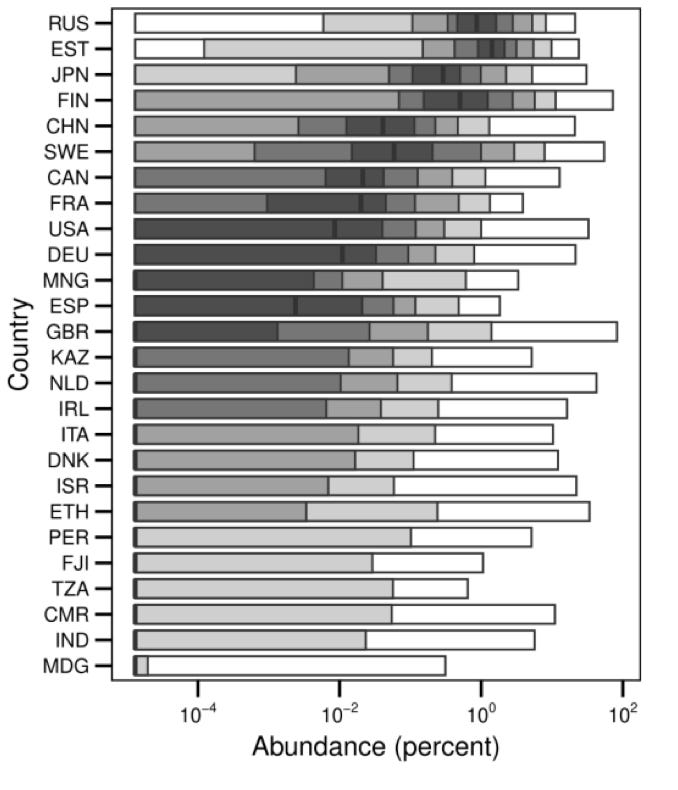
So this is how I picture it going: Say you’re in the gnavus+ half the population (there’s two kinds of people in this world, huh?), but you’ve got a moderately low relative abundance; maybe you’re the average American in that chart above, and gnavus makes up 0.01% of your microbiome. That’s a very minor component! For comparison, one species of Bacteroides—which I often describe as the trees of the forest—might be 10% of a person’s microbiome by cell count. But just to get a rough physical sense of how much this is: say there’s 200g of bacteria in your colon. 0.01% of that is 20 miligrams of cells.
Now say you take an antibiotic, and it happens to be one that your strain of gnavus is resistant to. And after that antibiotic, you find yourself in the white bar for the US on that graph: top quantile, with its low end at 1% relative abundance, and its top at about 10%. Now you’ve got between 2g and 20g of gnavus cells in your gut.
20 grams! Y’know what else weighs 20 grams? A prostate! A thyroid! And so suddenly, at some point in young adulthood, with a little bad luck and a class of drug that the average American takes once a year, you’ve gone from being the kid who gets a little weird when he eats too much pizza to being the guy who has grown a small organ dedicated to churning out psychoactive chemicals. A month or so after you’re off the antibiotic, your microbiome has reached its new equilibrium, and the switch has been flipped.6
In my parents’ day, the popular explanation for this sudden switch-flip was “hormones”; maybe something about the end of puberty activates it. And maybe “adolescent synaptic pruning” is the more evolved version of that, incorporating the fact that most of the schizophrenia risk genes worth talking about are immune system genes, and synaptic pruning has common heritage with some immune processes.
“If your microglia did a little too much pruning in the executive function department”, the thinking goes, “and then pruned the brake lines on the dopamine system…” you could get something that might reasonably be expected to look like schizophrenia.
But that still leaves a lot of explaining to do: how does that gene actually cause the bad pruning to happen, and why doesn’t it happen in the other 98.8% of the people who carry the gene? Or if it’s polygenic, how does that gene interact with any of the other genes involved to produce that effect? Because my main problem with the genetic hypothesis of schizophrenia is that practically all the risk genes identified to date…
(A) do so little to impact disease risk that you need a cohort of about 2,000 people to even identify them as being more common in schizophrenia, or
(B) have nothing to do with neurobiology, in terms of their known functions.
…if not both.
Saying “highly polygenic” or “gene-environment interactions” is not an explanation, it’s a description of an explanation that you hope to come up with someday. That kind of thing has slid by, thus far, because everybody at the table in these conversations is just as naked and perplexed as everyone else, a state that generally engenders polite agreement when the subject of fashion comes up. But the minute someone shows up with a simple, mechanistically valid, testable hypothesis and biomarkers to go with it? You gotta engage with the specifics of the idea!
If you want to argue that the effects of chronic phenethylamine and tryptamine infusion don’t resemble schizophrenia very much, be my guest—but then you have to contend with the fact that amphetamine psychosis is one of the only animal models of schizophrenia with any translational validity, meaning: the drugs that work to alleviate amphetamine psychosis also tend to work for the positive symptoms of schizophrenia.
In the comments, someone also raised that the hallucinations of schizophrenia are not much at all like those induced by serotonergic hallucinogens. Now, I have taken my fair share of tryptamines and phenethylamines in my day (arguably more than my fair share), and I think I agree with this person. At anything less than “breakthrough” doses, you don’t see things or hear things as much as you feel things, like a psychic high-pressure front that infuses every situation with a sense of ambient significance. As I understand it, the delusions and hallucinations of schizophrenia are much more like what happens to you after a few days of sleep deprivation: you glimpse the coat rack out of the corner of your eye and it makes you jump, because for a second you could’ve sworn it was someone standing there—and even after you calm down, you can’t shake the feeling of another presence there in the room with you. Or you’re dozing, and then startle awake at a noise out on the street—was that someone saying your name?
So consider that, if you put someone on a phenethylamine slow-drip, you’d expect them to sleep pretty poorly, if at all. Add in that feeling of impending…something you get from a tryptamine, and that sounds like a recipe for a pretty psycho time.
But I have never had schizophrenia—and neither, I suspect, has Scott Alexander. And because I don’t plan on Barry Marshalling this one unless I absolutely have to, I’d be curious to hear if any of our readers check both of those boxes, and can give us an actual firsthand opinion on to what extent one of those things is like the other.
IIIa. Flipping and Unflipping the Switch
Before we move on, this seems like as good a place as any to address one of the more thoughtful critiques that came up in the comments of “Contra Skolnick”, courtesy of a user by the name of Xcm, who asked pretty insistently: “Shouldn't a course of antibiotics to which R. gnavus is susceptible immediately result in noticeable improvement?”
And it’s a good question! But “we’d have noticed by now” is the kind of argument that often collapses immediately when you try to move it from the vacuum of imagination into the real world, with all its confounders.
Are we talking about a macrolide, or a beta-lactam antibiotic? Is it given by mouth, or IV infusion?
By mouth, the first tablet you give will maybe take 30 hours to reach the rectum, which is where it matters. If it's a protein synthesis inhibitor or something like flagyl, it won't do much to inhibit the activity of an enzyme that's already been produced, so you're probably waiting at least 2 defecations (somewhere between 25 and 80 hours) before things get any better.
Would we notice?
Well, was the person in active psychosis? That's when you'd expect an immediate improvement to be most noticeable.
But how often does a doctor prescribe an antibiotic alone to a schizophrenic patient in active psychosis, without also prescribing something to try and manage the psychosis? And if that thing seems to work, don't you keep them on it?
If they weren't in psychosis when you gave them the antibiotic, then yeah, maybe they see a durable remission in negative symptoms over the following months. So then we’re back to: why were we giving them the antibiotic? Say it's a UTI, and at 6 months post-drug, they say "hey doc, I've been feeling a lot better, no like a LOT better since then..." How much better do they need to be, and for how long, before you to write up a case report rather than saying "I'm very glad!" and chalking it up to clearing up the UTI?
The problem is that, in the short term, remission is nothing newsworthy, and in the long-term, bacteria grow back unless you take such a complete scorched-earth approach that it's liable to cause problems of its own. If you nuke it all and then replace it with an FMT, that’s one thing—but as far as I can tell, nobody has tried an antibiotic+FMT for schizophrenia. The one scientific case report I can find, of a person making a complete and durable recovery from serious schizophrenia with no antipsychotics, is the story of a young man who got a bone marrow transplant. The protocol for this involves so much chemotherapy that it literally hollows out your bones, followed by a whole new immune system and thirty days in a sterile isolation room, during which time the man’s psychosis disappeared, and stayed gone for at least four years. And because the chemo drugs involved in that kind of thing are toxic to anything that has DNA (i.e. everything), this is one of the few people who I’m confident actually got a fresh start on a microbiome. Confounders abound, of course, but we can at least be pretty sure that the bone marrow transplant didn’t un-prune his adolescent synapses, so there’s that.
Also worth noting: that paper was really hard to find, because—if you put “antibiotics” and “psychosis” into the same query on scholar.google.com, most of your first five pages of results is full of case reports, studies, and reviews about the problem of “antibiomania”—the fact that full-blown psychosis is a relatively rare side effect of nearly every kind of antibiotic.

Remarkably, this parallels the pattern in multiple sclerosis, where practically all antibiotics increase your risk, but the risk ratio depends really heavily on the type of antibiotic. It would be one thing if it were specific to a class of drug: aminoglycosides are famous for causing hearing problems, and in that case it’s something about their particular mechanism of action—their molecular structure, and the way it kills bacteria—that makes them liable to do collateral damage to the sensitive cells of the inner ear.
But most of the different antibiotics in that table look nothing alike structurally, and they work by totally different mechanisms of action. The only thing most of them have in common is that they kill bacteria.
And I didn’t even think to bring it up in my discussions with Xcm, but it’s really a solid corollary to their question. WOULD we have noticed by now if it were possible to treat psychosis with antibiotics? I don’t know. But we’ve definitely noticed that it’s possible to induce it.
IIIB. On Timing, and Aioli Problems
You ever had a really amazing mayonnaise?
Of course not. What would that even entail? Maybe there’s garlic mixed in, some spices? Well, that’s no longer mayo, is it? What you have there is an aioli, maybe a remoulade.
And I know it’s silly, but I think it gets to the heart of something that everyone knows is a problem in psychiatry, i.e. that the names we assign things can lead us to treat two things as the same when they’re quite different, or vice versa. I once saw a talk where an fMRI guy claimed that, if you break it down by what’s going on in the brain on their scans, the disease we call “major depressive disorder” could be as many as 12 separate phenomena.
But it goes the other way, too. One of the great mysteries of schizophrenia is its timing: why does it only seem to strike, as Scott put it, at ages 18-30? How do I explain that with bacteria?
Of course, it doesn’t. Just in my own life, that’s plain: two good friends of mine have siblings with it. One “grew into” her diagnosis: it started out as severe ADD and a learning disability at a young age; once she was old enough, she was transitioned to a diagnosis of schizophrenia without much fanfare or change in her medication regimen. The other got his diagnosis pretty young, compared to most: 13 or 14. Weirdly enough, that same friend’s mom had a bizarre amnestic episode in recent years, and apparently lost all memory of roughly the last decade. A stroke? Early onset dementia? We’re not sure. She was always the kooky mom, in our friend-group. Great fun, the perfect picture of “only slightly weird”, to use Alexander’s phrase.
Point being: Imagine for a moment it’s all as simple as I say it is, and schizophrenia is basically like being put on a semi-constant internal slow drip of psychoactive drugs, with the bonus that tryptamine appears to mildly fuck with your ability to synthesize proteins, because it looks too much like tryptophan. Now imagine that same process starting at age 2 or 3, while you’re still developing language skills. I mean, you can’t really imagine such a thing, because the brain of a child that age is so different from an adult’s, but we might expect them to stop learning new words. It might look a bit like some cases of ASD.
Or imagine it happening at age 70, when you’ve already got a full lifetime of experience navigating the world, the strong network of social bonds that younger-onset schizophrenics often don’t develop. What would that look like?
IV. Spouses, Siblings, Species, and Strains
One of Scott’s main objections to this hypothesis was that schizophrenia’s heritability patterns don’t look like what you’d expect from a microbiome-driven disorder—or rather, they don’t look like what he’d expect, because he’s operating under some false assumptions.

Here again, this is my fault for omitting some details in the name of brevity. Because, while the microbiome is certainly kept on a longer leash than most of the rest of our biology, the leash is still there—and in a lot of ways it does lead back to the genome. The main thing holding the other end of that leash is the immune system, but there are also major contributions from less-direct factors like mucins: amino-sugar-rich proteins secreted by your intestinal cells, which serve as both a barrier and a food source for certain bugs. Or think about lactose intolerance, or even the olfactory receptor genes that determine your sense of smell, your taste preferences. To the extent that these differ between fraternal twins, you’d expect to see more microbiome differences between them than you do with identical twins.
And if you plug some basic keywords (“microbiome twins identical fraternal”) into google Scholar, you’ll find that identical twins do, in fact, have more similar microbiomes than fraternal ones.

What’s especially neat about the above table is that they’ve even broken out the analysis phylogenetically, so we can see that—while monozygotic (identical) twins aren’t more similar than dizygotic (fraternal) twins with respect to the Bacteroidaceae in their microbiomes, they are more similar with respect to the Lachnospiraceae—which happens to be where Ruminococcus gnavus sits. Of course, that’s only by the “weighted UniFrac” metric, and I can hear the skeptics already: What, he’s just gonna pick the metric where there’s a significant difference in the taxon of interest? What about the unweighted UniFrac?
Well, first things first: UniFrac distance is basically a measure of “How different are these two microbiomes?” (UniFrac stands for Unique Fraction of the tree of life) but that’s a tricky thing to reduce to a single number, so you have to tread carefully when trying to figure out what a study like this is actually saying. Is a pine forest in Oregon more like an oak grove in Oregon, or is it more like a pine forest in Japan? It depends on what you really want to know about, but it’s worth learning how UniFrac works so we can at least talk intelligently about these findings.
For a simple case, imagine two valleys next to each other, and let’s say that they both have the same two species of tree in them: one oak and one pine. That’s a UniFrac distance of 0, because there’s nothing unique between them. But if we introduce squirrels to one valley and not the other, now there’s some UniFrac distance between them. Let’s arbitrarily say that’s a distance of 1. If we put a couple foxes in the other valley, the UniFrac distance between the two would go down, maybe to 0.5, because—while foxes aren’t that much like squirrels—they’re more squirrely than trees, which were the closest thing in the valley before the fox showed up. If, instead of foxes, we put an eagle in the other valley, the UniFrac distance would still go down, but not by as much: a bird is more like a squirrel than a tree, but less like squirrels than a fox. A chipmunk, on the other hand, is about as squirrely as you can get without actually being a squirrel. So if the two valleys both have the same species of trees, but one has squirrels and the other has chipmunks, we’d maybe have a UniFrac distance of 0.1.
So now take it back to the two-trees-only case, pines and oaks in each of these valleys, UniFrac distance 0. But say one valley is a 50/50 mix, while the other valley is all pines, except for one lonely oak. Now it doesn’t seem right to say “there’s no difference between these two,” right? That’s why weighted UniFrac also takes the relative abundance of the species into account, rather than just checking “what’s there?”
Knowing all this and taking it back to the table above, we can now see it’s telling us that—where the Lachnospiraceae are concerned—identical twins have more similar microbiomes than fraternal twins, when you give due weight to the ones that actually make up a significant portion of the ecosystem.
A beta-diversity metric like UniFrac isn’t really suited to investigating a hypothesis centered on a single gene or enzyme, like this is—it’s the kind of thing you look at when you don’t know what you’re looking for, which is why the points Scott makes below about spouses don’t really hold up. But it’s still a neat thing that what Scott thought was a “gotcha” turns out to supports the hypothesis.

Okay, to quickly address that last one:
Can we think of any reason why there might not be a lot of studies showing that children adopted out from mentally healthy parents to parents with schizophrenia acquire the schizophrenia risk of their adoptive families? Any reason at all.
This is the kind of objection that tells me that the guy gave just enough thought to my words to write a semi-coherent rebuttal, and even less thought to his own words as he wrote it. And this is the thing that pissed me off enough to elicit such a long and thorough reply.
It is right to be skeptical. It is a sin to give your skepticism the benefit of the doubt.
So let’s talk about the “epicycle” he describes in the first bit, where he’d expect the genes that determine your microbiome to show up on GWAS as schizophrenia genes.
If you’ve been reading attentively this section, you’ve maybe already put it together: I mentioned in the bit on synaptic pruning—practically all the major schizophrenia risk genes are immune genes! Mostly located in a region called the Major Histocompatibility Complex, or MHC. This is part of how the body differentiates its own tissues from those of other species which might be invaders. It’s not fully understood how the body compiles or enforces its list of which bacteria are friends vs. foe, but it’s all too easy to imagine that one of the genes in there is the molecular equivalent of a “DO NOT ALLOW ON PREMISES” poster, with a little picture of his glucorhamnan surface polysaccharide.
V. The Poison Makes the Dose
Probably the most consequential objection to this theory is the possibility that we've got the direction of causality wrong here. All the schizophrenic people in this study were on medications like clozapine, so how do we know that clozapine doesn't cause the observed microbiome differences?
And it’s true, the authors of the paper note that there’s a significant correlation between antipsychotic dosage and the abundance of bugs like R. gnavus, and even go so far as to conclude that it's probably the medication driving the observed microbiome differences—so I get why it seems shady that I didn’t mention the authors’ initial conclusion in my post.
The possibility that clozapine impacts the microbiome is worth thinking about, especially in light of the fact that it's known to produce constipation and other pretty serious GI symptoms. And, if the participants in this study had been randomly assigned to various clozapine dosages, then sure, the association between drug dose and gnavus abundance would be pretty solid proof that the drug drives the bug.
But that’s obviously not how this works: a patient’s medication dosage is a product of symptom severity—the more severe their schizophrenia, the more antipsychotic it takes to keep them out of psychosis. So there are two models here: in one, the symptom severity drives medication dosage, and medication dosage drives gnavus abundance. In the other, gnavus abundance drives symptom severity, and symptom severity drives medication dosage.

If you’re not a total shit-head like me, I can see why Model 1 would seem like the more likely possibility. Maybe you look at the top few rows in your results table, and go “huh, what is Faecalicatena gnavus, and is there any way it could drive symptoms?” and you do a lit search on it. Unfortunately, bacterial taxonomy is a fucking mess, so you’re probably not going to find the paper from 2014 where they characterized its aromatic amino acid decarboxylase, or this more recent deep-dive on the species’ pangenome, where they show that it’s a core feature of the species—because in both of those it’s referred to mainly as Ruminococcus gnavus. You’re also not going to be aware that Enterocloster—one of the other taxa most enriched in the schizophrenic gut—is one of the only other gut bacteria which possess such an enzyme; I only know this because we were looking into this pathway back at Holobiome and I read Fischbach’s original 2014 paper.7
But this is a key fact, because in light of it, it suddenly doesn’t seem like nearly as much of a stretch to suggest that the abundance of the bacterium is what drives the medication dosage. And if you want to postulate that it goes the other way around, you find yourself going “guess it’s a weird coincidence that a schizophrenia drug happens to facilitate the growth of a bunch of tryptamine- and phenethylamine-producing gut bacteria”. (Not THAT weird, because monoamines encourage motility, which probably contributes to a low baseline relative abundance of gnavus in people who aren’t otherwise constipated—but still.) Like synaptic pruning, it’s the kind of thing that makes sense to postulate in the absence of an explanation involving fewer unknown steps, but in light of what we know now, falls away to Occam’s razor pretty quickly.
VI. The Ball
If I may speak directly to you, Scott: I get why your first instinct was mostly to defend your framework rather than engaging earnestly with mine—to dismiss me on something like the MZ/DZ twins concordance before even checking whether it was true.
Because this game has been going on for centuries without a single point scored. When was the last time anyone even saw the fucking ball? For our entire lifetimes, it feels like everyone’s just been milling around the field looking for it, speculating about where we might find it, dreaming up plays for when we do. Does anyone remember what it looks like? How sure are we that there even is one?
So I understand the skepticism, when you hear someone shout—not too far off—I’VE GOT THE BALL!.
The false starts / true starts ratio, to-date, is ∞. And when you look over, and you see that the ball in question is one of these…

…you roll your eyes. This is a joke, right?
Yes, absolutely. If there’s one thing my artist friends are pretty clear on, it’s that all of this—the whole ballgame, you and me included—none of it makes sense except as a cosmic joke.
But that just would be the punchline, wouldn’t it? The one thing nobody’s tried, because until this very Sunday morning, it sounded…well, yes, crazy.
So I’m telling you: I’ve got the ball. I’m running it.
And if I can make it to the end zone, I guess we’ll see what happens. But we’re on the same team here, unless you’re playing for Moloch, so the only question is: Do you want to sit on the sidelines and watch, or do you want to get downfield so I can pass to you?
Because I am pretty good, but I’m only one guy. I need help. Someone with an MD. Preferably a psychiatrist. Because, unlike the thousand SNPs in the genome, this is an extremely testable hypothesis: a course of vancomycin and rebyota. Drugs that you could write a scrip for tomorrow.
So this is all I ask you: adopt this hypothesis for a minute; hold it in your mind as if it were your own. I do not mean be gentle with it; you must be violent! The whole point is that you try to break it: smash it, chip at it, find the seams and pry them open. Yes, ask the questions—If that were so, then wouldn’t we see…—but then you must actually try and find the answers! Dig into the metabolomics table, read the papers, check your assumptions—see it for yourself. And if you don’t just toss it away, saying “I’ve seen a dozen like this”, I think you’ll be surprised. Nothing I’ve tried yet has cracked it, which is why I’ve invited the world in to come and have a kick. And if it crumbles in your hands, I’ll thank you. We didn’t want that one anyway.
Because we are after the truth.
And truth is indestructible.
—🖖🏼💩
Huge thanks to everyone who engaged in the discussion around this in the comments of my last post and “Contra Skolnick”. A lot of new readers came from that one, a lot of new ideas. It’s a heartening reminder that—even if it turns out Scott’s now a replicant—the best thing about SSC/ACX has always been the community of people who gravitated toward his writing, because they recognized the spark in it. Y’all know what the real thing looks like.
Especially tricky in psychiatry, which probably attracts crackpots at a rate that outpaces even theoretical physics.
The same goes for “heritable”, which geneticists will insist does not actually mean “able to be inherited”.
I haven’t read their methods indepth, but this one’s probably gotten more “shots on goal” than any other indication. At present, it seems more likely to me that—along with the obvious problem of too many calories—the obesity crisis has something to do with the fact that the most widely used herbicides in modern agriculture are sulfonylureas, a class of chemical which includes a number of once-popular diabetes drugs. These work by triggering insulin release from the pancreas, and they aren’t used much for diabetes treatment anymore because constantly hitting the “release insulin” button leads to insulin resistance. (Stay tuned for the upcoming post exploring this in more depth.)
Yes, it crosses the blood brain barrier. Someone raised this to me recently as a general concern about microbiome metabolites, which is fair—but this is one thing we know pretty certainly does. Interestingly enough, in googling to find a citation for that, I learned that in animal experiments, tryptamine can induce a neurodegenerative syndrome which includes amyloid accumulation as a feature. Hm, maybe this will be important later.
Not to be confused with blueballs, which definitely exist despite what your mom might have told you.
At that point the rate-limiting factor on tryptamine and phenethylamine biosynthesis is likely the amount of those amino acids in the gut, rather than the abundance of the decarboxylase enzyme. (This is assuming that the main reason gnavus produces tryptamine is to keep other bacteria nearby from getting their hands on it. If tryptamine itself has growth-inhibitory effects on other bacteria which gnavus is immune to, then there’s a model where it would be worth it to gnavus to build the tryptophan from scratch, just to produce the “antibiotic”.)
When I joined, they were looking into it as a potential treatment for depression and maybe constipation, under the logic that an aromatic decarboxylase is one of the two steps that takes you from tryptophan to serotonin. One of the reasons I left was that I felt that the founder was too cautious—that we should be taking these things straight to human testing. In this particular case, it is probably a good thing that he didn’t listen to me, because we might have ended up giving someone schizophrenia if we had. Then again, if you know how to cause schizophrenia, you’re most of the way to knowing how to cure schizophrenia, so who’s to say?
> he used an LLM to write his rebuttal post, and this was sort of a “hallucination”.
The more I think about the latter possibility, the more likely it seems. It fits the pattern of confidently wrong assertion-with-citation that you get from current models, Scott just mentioned dealing with o3 hallucinations in the writing of a recent piece, and he seems like the kind of guy who would have a custom GPT trained on his corpus.
----
I believe he's said previously that he does this. I can't search my old comments on Substack, but I had a conversation ~6 months ago with someone after he issued a correction like "o3 said this so I included it, but it's actually not true at all, my bad" for something that was obviously not true if you spent more than ~20 seconds thinking about it. I'm really worried that AI is slowly replacing the necessity of struggling with hard or controversial concepts, and taking the benefits of that struggle: understanding, along with it.
The interesting thing about your stuff is that it actually seems testable in-vivo. Unlike other crackpot theories, which are usually unfalsifiable or make no clear hypothesis at all, it seems relatively straightforward to me, a layman, that you can just act on someone's gut microbiome to introduce strains of bacteria that aren't currently there. Add the cholesterol-eating bacteria (or whatever the mechanism is, I remember it increases cholesterol in urine or something like that), and see if someone's cholesterol decreases. If it does, then that's pretty strong evidence that the theory is sound, and repeated intervention over a larger group would slowly turn it from hypothesis to fact. I'm sure it's actually much more complicated than this, but whatever the specifics, it seems like there's a broad-strokes path to falsifiability, so your stuff is therefore quite interesting.
Re: whether psychosis feels like a psychedelic or an amphetamine, I don't have schizophrenia, but have bipolar I and have gone through mania/psychosis 3 times.
My mania *absolutely* resembles a psychedelic / stimulant experience - to the extent that the first time I had a psychotic break, for part of the lead-up I was convinced I'd been dosed with LSD.
Imagine being on a kind of euphoric psychedelic + ritalin (or what I'd imagine meth to be like) for days/weeks/a month straight, and completely losing touch with reality as the trip gets deeper- transforming into an agitated, 'bad trip' over time.
It's unique, but it doesn't feel categorically different. The closest analogue I have to early mania was when I tried DMT and didn't "break through," plus a stimulant.
The things that do stand out are the hyper-confidence, the grandiosity, the feeling of the conscious experience radically speeding up, and the partial memory blackout.