
Vitamin Q
or: Why You Literally Can't be Happy Without Bacteria

When I’m talking to someone new about the gut-brain axis, I’ll usually start by asking: “How’s your neuroscience?”.
In most cases, this gets a grimace, coupled with an “ehhhhh” noise, and the kind of hand-wiggling motion which—at face value—means “so-so”, but which in this context indicates “I have next to no idea how this thing on my shoulders works.”
So it’s usually with a relieved smile that they’ll answer my next question: “You know about dopamine?”
Oh, easy. Everyone knows about dopamine.
“And serotonin? You’re familiar with serotonin?”
More enthusiastic nodding—of course. We’re two for two!1
“Good!” I tell people. “You’ve already got all the background you need.”
Why and How
Whether or not they get all the details, nearly everyone is on solid footing with the understanding that these neurotransmitters are important for mood, memory, and the ability to think. Maybe you’re aware that, when people talk about SSRIs being the most widely used kind of antidepressant, one of the “S”-es stands for “serotonin”. If you’re a real overachiever (or an intellectually curious degenerate), you’ll be aware that drugs like cocaine and amphetamine work by activating the serotonin, dopamine, and norepinephrine systems.
Knowing that these drugs tend to impart energy, enthusiasm, and the feeling that everything is cool and froody—and that they do this by mimicking those three neurotransmitters—it’s all too easy to understand why it’s catastrophic if something goes wrong with your body’s ability to produce them. This is the “neurotransmitter imbalance” theory of depression, and—even though we finally got direct evidence recently that depressed people’s brains really do release less serotonin than healthy people’s2—this explanation still kicks the can down the road: It’s a how, not a why. If a neurotransmitter imbalance causes depression, what causes the neurotransmitter imbalance?
To get the obvious caveats out of the way: a zillion different things—from heavy metal poisoning to genetic mutations to infections—can mess with neurotransmitter biosynthesis. Talking about depression as if it’s a single, explicitly biological condition necessarily paves over a lot of nuance: Odds are that maybe 40% of people with the diagnosis “major depressive disorder” have one specific thing wrong with them, another 30% have some completely different issue which happens to result in similar symptoms, another 10% have some third thing, and so on. Add in psychological and social factors, and the picture gets even worse. The same is true of schizophrenia, bipolar, autism, and a host of other psychiatric, neurodevelopmental, and personality disorders. This is one big reason why none of these diseases are well-understood at the root-cause level yet.
But another reason is that practically everyone trying to understand these diseases over the last century has been starting from the premise that—if we want to learn about them—we should be studying the brain. That work has turned up a number of leads and insights: differences in the size or connectivity of one lobe or another, neuron growth factors like BDNF being higher in healthy controls, and the famous neurotransmitter imbalance…but the closest we’ve come to a why are things like “oxidative stress” and “inflammation”, both of which leave a lot to be desired by way of actionable solutions. (“More exercise, more sleep, and uhhh, drink green tea?”)
In recent years, though, we’ve begun to see solid evidence that—although the how certainly unfolds above the shoulders—the why might lie a little lower down, in the gut microbiome.
It’s an intuitively pleasing idea for a lot of reasons, from truisms about “gut feelings” to the well-established associations between diet and mental health—but first and foremost is that the microbiome is the one place in human biology that we haven’t really looked yet. The genome has been checked pretty thoroughly, and for the most part ruled out: there’s no reliable way to tell whether someone has depression or schizophrenia just by looking at their genome. This fact has been driving scientists batty for decades, because most of these diseases ARE very clearly heritable: if you know whether a person’s parents or siblings have a mental illness, you’ll have about ten times better luck predicting their mental health status than the best genomic analysis can do.3
This discrepancy has led some scientists to theorize that there must be thousands of different genes involved, each of which influence the risk of mental illness a tiny bit—making the impact of any one hard to suss out. But over the last ten or twenty years, a much simpler explanation has emerged, with the discovery that the gut microbiome is not only highly heritable, but wildly variable across the population—and even within a given person over the course of time.
That heritability helps explain the genome discrepancy, and—more importantly—it makes it seem a lot more plausible that the microbiome could be necessary for key biological processes. Before we understood that core components of the microbiome are passed down as reliably as any of our own genes, it seemed unlikely that we would rely on gut bacteria for critical things like “staying sane” or “wanting to live”.
But sure enough, as the technology has gotten good enough for us to just shit directly into a gene sequencer and see what’s going on in there, we’ve found that the gut microbiome is out of whack in a number of psychiatric diseases—and that here, too, you can do much better at predicting whether someone is mentally healthy vs. ill than you can by looking at their genome. This is extremely exciting for two reasons:
First off, it offers the prospect of an ultimate, simple, satisfactory why. A disruption to your interior ecology is to blame—some bacterium that’s there where it shouldn’t be, or missing where it was present in every one of your ancestors. We’ve all taken antibiotics at some point in our lives, which is a little like playing Russian roulette with the gut’s ecosystem: Most of the time, nothing bad happens—but there’s always a nonzero chance that you’ll drive an important species extinct. If it turns out that species was essential for helping you make dopamine or serotonin, maybe you end up with depression.
That’s a simplified example, but it brings us to the second reason this is exciting: the microbiome is also infinitely easier to modify than the genome. If we find a bacterium that’s absent in some people with depression, but present in everyone healthy, replacing it might just fix things for those people.
So here we find ourselves “kicking the can” from the other direction: with a plausible why locked in, we can look for how something wrong in the microbiome might lead to a neurotransmitter imbalance.
The better part of a decade ago, I went out looking for that how, and I found one. A direct route from the microbiome to the monoamine neurotransmitters—serotonin, dopamine, and norepinephrine. No hand-waving, no unknown mechanisms: a clear hypothesis, and—if it turns out to be true—a simple solution. This is the idea that made me quit my job as a science writer, file a patent, move to Boston, and dive headlong into shit, so to speak. An idea so compelling in the scope of its implications that, even if there were only a one-in-ten-thousand chance of it being correct, it would be worth reorienting my entire life to try and find out.
This is the Vitamin Q Hypothesis.
What Could Go Wrong?
The catalyst for this grand idea, which hit me in about 2016, was a scientific article from 2011. The moment I read the title, I was thunderstruck—it was suddenly right there, clear as day—and by the time I’d finished reading the abstract, it took everything I had not to jump out of my seat in the café and shout eureka. We’ll get to that article and its magic title in a little bit, but we’ve got some ground to cover first before it’ll truly make sense.
So let’s start with the basics: serotonin and dopamine. Chemical cousins, they look somewhat similar, but they’re independent of each other: Each lands on its own unique set of receptors, which do different things in different parts of the brain. Serotonin doesn’t activate the dopamine receptor, and vice versa. Nevertheless, drugs that activate one often activate the other, thanks to their structural similarity.

That resemblance is no coincidence: although each is produced in the body from a different amino acid (serotonin comes from tryptophan, and dopamine comes from tyrosine), those parent amino acids are both produced by a set of molecular machinery referred to as the shikimate pathway. Humans don’t have this machinery, so tryptophan, tyrosine, and the related amino acid phenylalanine, are all considered essential amino acids: our bodies can’t make them from scratch, so we have to eat them (although the microbiome probably kicks in some—see the Thousand Secret Ways).

OH!
Now, even if you’ve never taken a chemistry course, maybe you can already see the important thing here: that these two critical neurotransmitters, dopamine and serotonin, are produced by a perfectly parallel process. It’s the same set of two simple transformations, carried out on two different starting materials.
The addition of a hydroxyl (OH) group turns tryptophan into 5-hydroxy tryptophan, or 5-HTP. This is the immediate precursor to serotonin.4 Installing an OH group in a similar spot on tyrosine turns it into L-DOPA, which is the precursor to dopamine. Sharp-eyed readers might have noticed that the second step in the diagram above—the chemical reaction that turns 5-HTP into serotonin—is also basically the same as the one that turns L-DOPA into dopamine: the removal of a CO2 from the “teeth” end of that key—a decarboxylation.
We’re not going to focus on that, though, because it’s the first step—the hydroxylation—that’s the important one here. This is the hard part for your body, the rate-limiting step in the synthesis of serotonin and dopamine in your brain: the cap on how much of these chemicals you can produce. And because you need serotonin in order to produce melatonin (and dopamine to produce norepinephrine and adrenaline), that chokepoint has the potential to impact a cascade of processes from mood and cognition to sleep.
That same hydroxylation reaction, it’s worth noting, is also the key step in preventing the amino acid phenylalanine from being neurotoxic: Because it looks a lot like tyrosine and tryptophan, phenylalanine can block up the transporters that are supposed to carry tryptophan and tyrosine from the bloodstream into the brain—leaving your neurons starved for serotonin and dopamine. Phenylalanine is abundant in most high-protein foods, but in a healthy body, an enzyme in the liver hydroxylates most of it into tyrosine before letting it into circulation. If that enzyme doesn’t work properly (some people are born with a “broken” version, thanks to a genetic mutation), you get a disorder called phenylketonuria, or PKU—where you have to religiously avoid phenylalanine, or else you end up with social and intellectual disabilities, a weirdly pale complexion (tyrosine is also the precursor to melanin), and a “musty” odor radiating off you.
All this to say: if we want to understand why some people seem to have a hard time sleeping at night, waking up in the morning, focusing in class, or just generally feeling like a human being ought to, it’s worth taking a close look at that rate-limiting step—at the little molecular machines that grab a tryptophan, or a tyrosine, or a phenylalanine, and stick an OH onto it.
These are the aromatic amino acid hydroxylase enzymes (or “AAAH”, if you like).5 There are three of them; a dedicated one for each of those three amino acids. Tryptophan hydroxylase turns tryptophan into 5-HTP, phenylalanine hydroxylase turns phenylalanine into tyrosine, and tyrosine hydroxylase turns tyrosine into L-DOPA.

Got it? Good.
Batteries
These three enzymes are pretty similar to one another, both in structure and in function. They’re almost identical, in fact—which means that all three run on the same fuel, a molecule called tetrahydrobiopterin.6 We’ll call it BH4, for short.
If these three AAAH enzymes are molecular machines for making neurotransmitters, BH4 is the battery that powers them: it’s a cofactor for the enzymes, meaning they can’t work unless they’ve got a molecule of BH4 attached in the right spot. Right off the bat, this seems like an important molecule, doesn’t it? Sitting at the corner of serotonin street and dopamine drive? Where do we get more of it?
Truth be told, you don’t want more biopterin. Your body can make BH4 from scratch, but making it isn’t the hard part.
Just like a battery, BH4’s charge runs down when it gets used: Every time your body converts a molecule of phenylalanine to tyrosine, or tyrosine to L-DOPA, or tryptophan into 5-HTP, a molecule of tetrahydrobiopterin gets “burnt up” in the process—it oxidizes to dihydrobiopterin, or BH2.

BH2 is like a dead battery in the junk drawer. It looks enough like BH4 that it can still slot into the right spot on the AAAH enzymes, but it just sits there—no juice, no serotonin. Eventually, that molecule of BH2 will drift off, and then there’s the chance to try again—but there’s nothing stopping another molecule of BH2 (or even the same one!) from drifting back in. I guess your body, in this analogy, is the kind of degenerate who will take a battery out of the drawer, try it in the remote, and—if it doesn’t work—throw it back in the drawer before grabbing another to try.
The important thing to understand about this dynamic is: how well the AAAH enzymes do their jobs is less about “do you have enough batteries in the drawer” and more about “what percent of these batteries have any charge?”
Now, your body has a good reason for keeping those dead batteries around rather than just throwing them out: you have enzymes that recharge them, reducing BH2 back to BH4 and making it usable again. Recharging the oxidized cofactor, rather than making a new BH4 from scratch every time you need one, is more biologically cost-efficient (imagine how much you would spend on batteries if your smartphone ran on single-use AA’s) but it’s also a logistical necessity, thanks to the “junk drawer” nature of the cell, and the semi-random way the AAAH enzymes bind their biopterin cofactor: the more batteries you have in the drawer, the harder it gets to improve your odds of getting a good one by adding in charged ones.
So there’s a cycle here, between the AAAH enzymes that oxidize BH4 to BH2—churning out tasty neurotransmitters in the process—and dihydropteridine reductase, the enzyme that reduces the BH2 back into usable BH4. At any given time, you have both forms of biopterin in a cell, at some ratio that varies depending on the relative activity of the enzymes that use up BH4 and those that regenerate it.

There’s a third factor here though, because BH4 is also very susceptible to nonenzymatic oxidation—meaning any free radicals floating around the inside of your cell can also oxidize it to BH2. This is part of how certain heavy metals (lead, in particular) mess with cognitive and psychiatric health: The concentrations of these all these factors in your tissues are such that anything which pushes the ratio of BH4/BH2 towards BH2 will slow down production of 5-HTP and L-DOPA, while anything which pushes it towards BH4 will speed it up.
And the same way that a paperclip in the junk drawer can short out a battery and drain its charge, reactive metals like lead can oxidize your BH4 to BH2 without doing anything useful—making it harder for the reductase enzyme to keep up. Lead isn’t unique there, though; iron can do the same thing if it’s not contained in a structure like heme.
And the same goes for any reactive oxygen species, like hydrogen peroxide generated by the immune system—or even the everyday byproducts of cellular metabolism that can build up if a cell is overtaxed.

When everything’s working properly, free-radicals are kept in check by antioxidant enzymes like thioredoxin and molecules like vitamin C or glutathione, and oxidized BH2 is enzymatically recycled into BH4, leaving you a steady supply. But the complexity of these dynamics is why, at the beginning of this section, I said “you don’t want more biopterin”: taking supplemental BH4 doesn’t really help increase levels of BH4 in your neurons, because most of it gets oxidized before it’s transported into cells—meaning you end up with a whole lot of excess BH2, which actually inhibits the enzymes we want to encourage. The hard part isn’t making enough BH4: it’s keeping it in the right oxidation state.
Fragile By Design
All this seems like a bit of a design flaw, no? Wouldn’t it be better if our mood and cognition didn’t depend on a molecule that’s extremely susceptible to random degradation?
Well…no. Think about the dynamic this sets up: it makes mood into a sensitive barometer for oxidative stress. It means that, if your cells aren’t happy at a molecular level, you’re not happy at the macroscopic level.
You can imagine the benefit this would have in an early-human society. Say your tribe moves into a new area; there’s lush vegetation and abundant fruit, plenty of wild game, and a freshwater spring. Maybe you wonder: Why has nobody else settled here? Did we just luck out?
But suppose the answer is No, because it turns out that the soil here is very high in lead—so the fruit, the game, and the water are full of it. In a world where serotonin and dopamine biosynthesis are independent of oxidative stress, you might not notice anything wrong until a year or so later, when the stillbirths start, and the kids who survive come out with strange defects. By that point, your whole tribe has accumulated enough of this cellular poison to increase their risk of cancer, infertility, etc. Even if you do move away, your elders will die early, taking their wisdom with them.
But in our world, where the serotonin and dopamine synthesis pathway is so exquisitely sensitive, the signs that something is wrong start to show almost immediately: The elders can’t sleep. Husbands bicker with their wives. Children cry inconsolably over nothing. It doesn’t take long for your people to conclude: this land is cursed, we must go. In this example, it’s clear that depression is not a maladaptive trait—it’s your body’s way of letting you know that something subtle is disastrously wrong, and you need to make a change.
But when the thing that’s wrong is internal, rather than external, it’s not as simple as moving to a different neck of the woods.
A Few Protons Makes All the Difference
Now, I am not the first person to recognize that biopterin issues are a promising candidate to explain the symptoms of certain mental illnesses. It doesn’t take a genius to look one or two molecules upstream of serotonin and dopamine and go “hey, what about this thing?”
But if you search the literature for “tetrahydrobiopterin+depression” or “tetrahydrobiopterin+schizophrenia”, you’ll find something curious: a preponderance of studies reporting elevated levels of biopterins in blood and urine samples from patients with these diseases. This, it seems, is enough to convince most researchers that this path is a dead-end. If someone’s already tested that, and the result was actually the opposite of what you expected? The issue must lie elsewhere. But if you read the materials and methods sections of these papers, you’ll notice a pattern: none of these studies are actually measuring BH4.
See, as we discussed earlier, there are multiple forms of biopterin in the body, and BH4 is sensitive to spontaneous oxidation: it doesn’t take much for it to lose a few protons and become BH2, or BH0. Oxygen, light, and time are enough to do it, at room temperature—so if you want to actually measure someone’s urinary BH4 levels accurately, you have to have them piss directly into a cup pre-loaded with three different kinds of antioxidants, in the dark, and then put the sample directly into a deep-freeze within a few minutes, until it’s ready for analysis.
This is a pain in the ass, so practically nobody does it. Instead, the protocol used in all of these papers goes something like:
-Collect urine sample as normal
-Treat it with iodine to deliberately oxidize all the biopterins to BH0
-Analyze to quantify biopterin
-Consult the literature to find the proportion that’s normally present as BH4 vs. BH2 vs. BH0.
Put simply: most people trying to investigate this have traded rigor for convenience, precisely at the most important point, because they aren’t giving the complexity of the system due consideration. You can see the evidence of this in the fact that—in the one or two studies that have taken the care to do it properly—you find higher total biopterins in the urine, but significantly lower levels of BH4 in depressed people than in healthy controls.
The same is true in schizophrenia: the BH4/BH2 ratio is all out of whack. This might come as a surprise if you’re only familiar with the disease from popular depictions—which tend to focus on the so-called positive symptoms: delusions, hallucinations, paranoia, etc. But in the vast majority of patients, these are accompanied by a suite of negative symptoms, which look a lot like a severe case of depression: emotional flatness, the inability to find pleasure in things, executive dysfunction, social withdrawal, and so on.
To make matters worse, the most effective drugs for managing schizophrenia’s positive symptoms are dopamine antagonists, which—as you might expect—make people feel like shit, and make the negative symptoms worse. This is why a lot of neuroscientists talk about the “dopamine paradox” of schizophrenia: the negative symptoms are characteristic of a deficit of dopamine and serotonin, but the positive symptoms can be quelled by drugs that prevent dopamine from doing its job.
The most plausible resolution to this paradox involves another molecule, one called kynurenic acid.7 This is one of the things that tryptophan can turn into, in your brain, if it doesn’t turn into 5-HTP first.
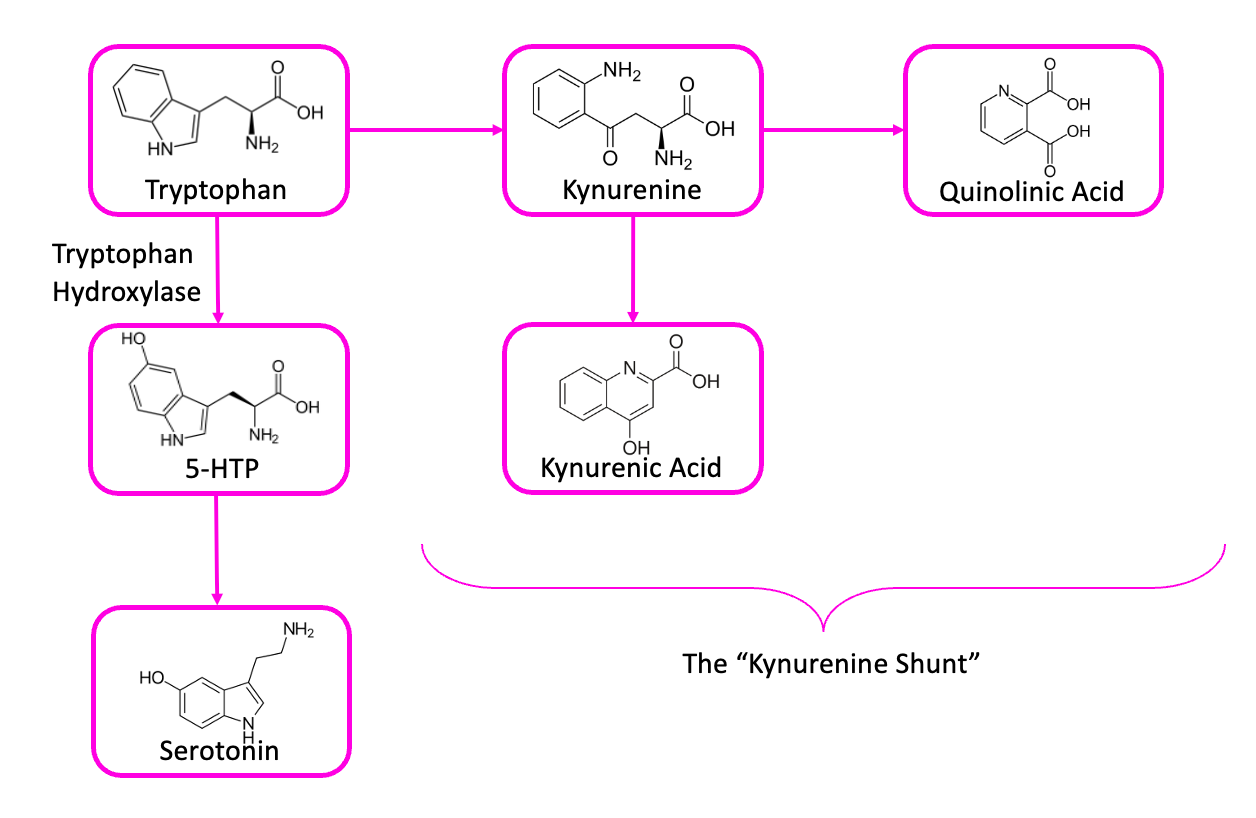
Kynurenic acid levels are higher in people with schizophrenia, which is particularly interesting because the molecule acts as an antagonist at the NMDA receptor—putting it in the same pharmacological class as drugs like PCP or ketamine, which are known to cause delusions, hallucinations, and the sensation that you’re really just a spinal cord piloting a meat suit. And because NMDA antagonists can cause dopamine-producing neurons in some portions of the brain to fire frantically, this is one of the best-accepted explanations for the dopamine paradox: some cellular defect impairs the production of both dopamine and serotonin, leaving your brain’s tryptophan with nowhere to go, except down the path that leads to kynurenic acid. Since kynurenic acid acts partly by causing hyperactivity in certain kinds of dopamine neurons, dopamine antagonists would be useful in preventing its effects.
That Magic Title
Okay, we’re ready. If you’ve been paying attention, you now have all the knowledge about this that I had when I punched a few key words into Google Scholar and had my life changed forever.
It was a simple query; something like “gut bacteria tetrahydrobiopterin”, and right there on the first page of results was this paper from 2011:
Queuosine Deficiency in Eukaryotes Compromises Tyrosine Production via Increased Oxidation of Tetrahydrobiopterin.
Now remember, the body ordinarily converts most of the phenylalanine you eat into tyrosine, with the help of one of the AAAH enzymes—which run on BH4. So this paper is reporting accelerated conversion of BH4→BH2, severe enough that it prevents the AAAH enzymes from working properly.
But what—I thought to myself as I clicked the link—the fuck is queuosine? How is that even pronounced?
Queuosine (“cue-uh-seen”, as it turns out), is a micronutrient that’s found in every cell of a healthy human body. All eukaryotes use it, but only certain bacteria can produce it. Plants (being eukaryotes) get it from their soil bacteria, and animals get it from their gut bacteria—or from the plants and animals they eat. In that sense, it’s basically a vitamin: something that your body needs in order to function properly, but which you can’t make on your own.
Unlike the better-known vitamins, queuosine was only discovered relatively recently—sometime in the 1970s, when scientists found that certain kinds of tRNA (the “adaptor” molecules that read the blueprint of mRNA and turn it into proteins) had an unusual modification that caused them to behave differently in chemical tests. Not knowing what they were seeing, the researchers reported on this as-yet-unidentified “Q factor”. When it turned out to be a heavily modified version of guanosine—one of the four standard nucleosides that make up the rungs of the ladder-like structure that is DNA—they kept the Q, and named it queuosine by analogy.

It would be a while before they figured out what it does, and longer still until they figured out that we can’t make it ourselves, but to pack a decade or two of research down into a couple of sentences: Q is grease for the gears in the machinery that makes proteins. It sits in a part of the tRNA called the anticodon (the bit that sticks to the mRNA) where it helps that sticking happen efficiently, and—just as important—helps the tRNA “unstick” when it’s done its job and added an amino acid to the growing chain of a protein.
Our bodies have enzymes that break down RNA from the digestive tract, freeing up queuosine from food or gut bacteria. That queuosine is then broken down into a smaller molecule called queuine—the nucleobase that corresponds to the queuosine nucleoside, the same way that guanine corresponds to guanosine, adenine to adenosine, etc.

Queuine is transported via the bloodstream to cells throughout the body, where another enzyme takes it and inserts it into the tRNA in the correct spot—swapping it out for a guanine. tRNAs that are supposed to have a queuosine in them but don’t can still do their job—but they don’t do it as well as they’re supposed to.
The upshot of all this is that, in an animal that’s queuosine-deficient, a number of proteins don’t get synthesized as fast as they should, and certain cellular processes start to malfunction. It took a long time for someone to actually test this in an animal to find out what goes wrong, because inducing a deficiency of something that’s produced by common gut bacteria is a pain in the ass.
See, ordinarily, when scientists want to study something like a vitamin E deficiency, they’ll take an animal, put it on a chemically defined diet—pure fat, sugar, protein, minerals, etc.—and add in all the known vitamins except E. When the animals start to look like they’re having a rough week, maybe you add vitamin E back in to half of their diets. Whatever this seems to fix, you can conclude is dependent on vitamin E. To do the same thing with a compound produced by gut bacteria, you need to start with germ-free mice. (Think “Bubble Boy”: born by C-section, doused in antibiotics for the first few weeks of life, kept in sterile isolators, and fed a diet that’s been gamma-ray sterilized to avoid introducing incidental bacteria or fungi that might take root in their guts.)
Because these experiments are costly and resource-intense to run, it was only in 1997 that someone actually managed to run a Q-depletion experiment—but what they found was startling. On a normal diet, germ-free Q-deficient mice didn’t show any overt abnormalities—or at least, none that could easily be distinguished from the abnormalities found in all germ-free mice. (Germ-free mice are not, generally speaking, a happy bunch. They are dumber and less athletic than their ordinary counterparts, and less social to boot.) But when they took the tyrosine out of these Q-deficient animals’ diets, every one of them died within a couple of weeks.
The reason, as that 2011 paper would eventually reveal, is that Q deficiency causes a failure in the enzyme systems which prevent BH4 from oxidizing into BH2. Too much BH2, and dietary phenylalanine stays stuck as phenylalanine, rather than turning into tyrosine. The researchers behind the 2011 paper even note in their discussion section that Q deficiency “could potentially limit the production of numerous biogenic amino neurotransmitters whose production is also under the control of BH4”, but seemed content to leave the investigation of this possibility to someone else.
Of course, mice aren’t humans, so it’s not a guarantee that a Q deficiency in humans would look like what we see in mice—but in this particular case, there’s good reason to expect that the effects of a Q deficiency would actually be worse for a person than for a mouse. See, one of the major differences between our species is that humans can’t produce ascorbic acid—AKA vitamin C—on our own, whereas mice can. Ascorbate plays a major role in the biology of biopterins, helping stabilize BH4 against spontaneous oxidation and keep it active, which means that humans ought to be more susceptible than most animals to things that disrupt biopterin’s sensitive oxidation-reduction cycle.
So, to recap: It’s been proven that, in mammals, Q deficiency leads to an excess of BH2, the “dead battery” which prevents the AAAH enzymes from working as they should. We know that the AAAH enzymes are essential for the production of dopamine and serotonin, which means that a Q deficiency should lead to a deficit of these neurotransmitters and their metabolites, accompanied by elevated levels of alternative byproducts. In other words: a deficiency of this microbial metabolite would lead to a syndrome that looks an awful lot like depression.

So we have here a single-molecule, vitamin-deficiency-style model of one of the most devastating mental illnesses in the modern world.
In Vivo Veritas
But how likely is it that you’d see a Q deficiency in humans? After all, if it’s such a pain in the ass to create a deficiency in animals, you’d expect it to be equally difficult in a person—especially since there’s no such thing as a “germ-free” human. Even if someone had a deficiency, shouldn’t you be able to fix it by eating enough bacteria? We’d have noticed by now if yogurt cured depression.
But here’s the thing: not all bacteria produce queuosine. Lactobacilli and Bifidobacteria—the two genera that comprise the vast majority of fermented foods and probiotics on the market today—only have the final few steps of the Q biosynthesis pathway in their genomes, which means they can’t produce it without help from other microbes.8 What’s more, a number of bacteria (primarily pathogens, but some “pathobionts” like Ruminococcus gnavus) have a Q salvage system much like our own, meaning they act as a “sink” for Q supplied by the diet or by other gut bacteria. The same is true of fungi like Candida. If you want to guarantee a Q deficiency in an animal, germ-free is the way to do it—but it’s entirely possible that some people’s microbiomes have an imbalanced ratio of Q producers to consumers, and that over months or years this leads to a deficiency in their cells. By analogy, we can look at vitamin K—another nutrient that’s found in food and is produced by gut bacteria, but which can be depleted by antibiotics. As it turns out, 8-31% of adults tested are found to be vitamin K deficient—so a Q deficiency seems plausible.
As of right now, the hypothesis that Q deficiency is involved in psychiatric diseases is perfectly valid—meaning that it all works logically: Q is essential for keeping up a healthy BH4/BH2 ratio, that ratio is essential for serotonin and dopamine production, and those neurotransmitters are essential for mood and cognition. Whether or not it’s true remains to be seen; that can only be determined through experiment, and in that respect this hypothesis is still almost entirely untested. There’s no data on the prevalence of Q deficiency in humans, or whether deficiency is associated with any particular disease.9 We don’t know how much a healthy person needs in a day, or how that figure might change with things like antibiotic use. We don’t even know how much people get from their diet vs. their microbiome; the few datapoints we have on levels of Q in food show that a lot of foods have next to none, while some (coconut water and wheat germ) have up to a few hundred nanograms per gram.
I mentioned at the beginning of this post that I filed a patent around it—a “use patent”, covering the use of Q to treat neuropsychiatric diseases, since you can’t patent a natural molecule. That patent still belongs to my old company Holobiome, and they’ve been working on the problem—but the methods we were using for characterizing Q levels there were laborious, and required hunks of liver tissue, which means we were restricted to working with postmortem samples of dubious quality.
But I suspect there’s an easier way. Queuine, the “free” form of the molecule found in blood, is a lot like the B vitamins, in that it’s water-soluble—which means anything in excess of what your body can use in a day comes out in the urine. Testing this hypothesis might be as simple as checking urine levels in a few dozen patients and healthy controls.
If you struggle with depression and you’re anything like me, you’ve already tabbed away to google “queuosine supplement buy” or something similar. If so: welcome back, dear disappointed ones. At present, there’s no company selling anything more than research-scale quantities (i.e. $500 for a miligram, only shipping to laboratory addresses).
But fortunately, you know a guy.
See, a year or two ago, I got fed up with waiting for preliminary data and went full mad scientist. I hit up a contract synthesis company and had them make a small batch of queuine, and (after having it NMR’d and mass-spec’d by a third party), I tried some. Unfortunately, I don’t have depression or schizophrenia, and the thing about vitamins is that you only really feel something from them if you were deficient in the first place. To the guy whose gums are bleeding from scurvy, a glass of orange juice is probably better than sex—but to everyone else, it’s just a glass of juice.
But I tried it, and when I didn’t die, I gave it to some friends—and we can now say that, at the very least, queuine is safe for human consumption at doses well in excess of what it should take to fix a deficiency.
One of those friends has a sister, though, who’s been suffering from schizophrenia her entire adult life. And when she tried it, there were some remarkable changes: her family described her as more engaged and helpful, more sociable. Apparently, after a few weeks she was feeling so good that she decided to go off her other meds. This, of course, caused problems of its own, but I consider it a promising data point.
The custom synthesis cost a little over $10k, and I’ve almost run out of my personal stash, but I’ve still got contacts at the company that produced it—so if you’re interested in trying some of this stuff and have a few grand to kick in on a project like this, shoot me an email and we’ll coordinate something. Doing the urinalysis discussed above would require “heavy” standards of queuine and queuosine—versions with an extra neutron in the nitrogens—and I’ve got a quote from an academic chemist who can produce these for somewhere in the neighborhood of $20k.
Cretins
If your only context for the insult “cretin” is from its use in Monsters, Inc., you might be surprised to learn that it was originally a medical term; it referred to someone with congenital iodine deficiency. Lacking the thyroid hormones that contain iodine as part of their structure, people suffering from the condition would end up short in stature, with enormous goiters, rough skin, and severe intellectual disabilities. As recently as the 1800s, you’d see entire villages affected by it to one degree or another in landlocked places with iodine-poor soil like the Alps.

And people in these villages just accepted this as part of life: some portion of the population, they must have said to themselves, is just gonna turn out as idiot dwarfs. What can ya do about it?
This turned out not to be true, of course. With the recognition of iodine as an essential nutrient, and the introduction of iodized salt over the following century, the disease vanished within a generation—and the insult started to lose some of its sting.
So this is my hope for the future: that in a hundred years, “schizophrenic” will be like “cretin”, or “scurvy”. Once, these words meant terrible things; they were the names of demons that might curse your child, or steal your life on a long sea voyage. Those words were full of ominous power, and we robbed them of it.
If a Q deficiency is truly behind depression or schizophrenia, we might be able to do the same, simply by adding it to the salt, or the flour—the way we do with the other B vitamins. And that might be all it takes to lock these terrible demons away in the annals of history—to turn their names into anachronisms, words without any real salience, except maybe as an insult that nobody can truly get offended by, or a word that children shout, playing pirate games.
Only time and data will tell if a Q deficiency really is involved in these diseases. It’s perfectly possible that it’s not; there are probably a dozen other things just like this in the microbiome, other valid hypotheses that could explain these diseases, waiting to be discovered and tested. But if this whole saga has taught me one thing, it’s that—even if this particular idea doesn’t turn out to be the one—there’s hope in the microbiome.
It doesn’t have to be like this.
—🖖🏼💩
If, at this point, they look like they’re feeling too cocky to learn, we’ll jump to “how about tetrahydrobiopterin?”
Although the neurotransmitter imbalance theory has been around for nearly as long as we’ve known about neurotransmitters and receptors, it took until very recently to conclusively show that depressed people’s brains release less serotonin than healthy people’s, because it’s tricky to sample someone’s brain chemicals without drilling a hole in their head—and even then, neurotransmitters start to degrade almost instantly when you disrupt the cellular structures that produce and transport them inside the brain. ANY study where they pull out or dissect an animal’s brain to look at levels of dopamine, serotonin, etc. should be viewed with extreme suspicion; to get accurate numbers that way, you basically have to use a rodent-sized guillotine and catch the head in a bucket of liquid nitrogen. In vivo microdialysis studies—where they implant real-time sampling tubes directly into the brains of living animals—are a little more reliable, but even these have their pitfalls, and none of the animal models for depression accurately reflect what’s going on in a human with the disease; if we knew how to produce the actual disease in an animal, we’d know how to cure it.
Scientists who study the genome tend to use “genetic” and “heritable” completely interchangeably, as if they’re the same word, which I think obscures some important nuance. The microbiome is obviously heritable, but so are things like your preferred brand of dish soap—if you grew up in a palmolive household, odds are your kids will too.
Which is why some people take it as a day-after supplement to counteract the “low” following a serotonergic drug like MDMA.
“Aromatic”, here, doesn’t mean “smelly”; it’s a chemistry term that refers to the pattern of double bonds in the ring structure. It is, of course, mere coincidence that the acronym for these enzymes—which are necessary for the biosynthesis of adrenaline—happens to be the same as the noise you make when adrenaline is released into your bloodstream.
You’ve heard of tetrahydrocannabinol? Well, this is…nothing like that, unfortunately, except I guess in the sense that it’s got four hydrogens on it somewhere.
Etymologically, a contraction of “canine urine”, because it was originally isolated from dog piss. Not to be confused with urocanic acid, named for the same reason, because I guess the early days of analytical chemistry involved a lot of dog piss.
There is one paper from the past year or so reporting the full Q biosynthesis pathway in a strain of Lactobacillus. This paper is a textbook case of scientific fraud in the modern era, and I’ve emailed the journal to see about having it retracted.
I have glossed over the absolute MASS of literature implicating queuosine deficiency in cancer, because—while I might be able to get away with telling you we’re gonna cure depression with a single molecule—I recognize that “…and cancer, too!” smacks of snake oil.
Oh my God, a new post! Had been worrying you'd dropped off the face of the Internet and I'd never get more non-stupid microbiome content. It's a niche to be sure, but I find your writing doubly relevant as someone who stocks groceries for a living. It's good to have a properly-calibrated level of cynicism about the products I'm pushing without going all RFK.
Apparently B. subtilis of nattō fame (https://stephenskolnick.substack.com/p/natto-king-of-fermented-foods) is an avid Q de novo synthesizer, relying on it for film formation and sporulation:
https://pmc.ncbi.nlm.nih.gov/articles/PMC10570037
Note when the study says “Absence of Q impairs sporulation and biofilm formation in B. subtilis” that absence was induced by a mutation, not by deprivation of environmental Q.
Nattō wins again?